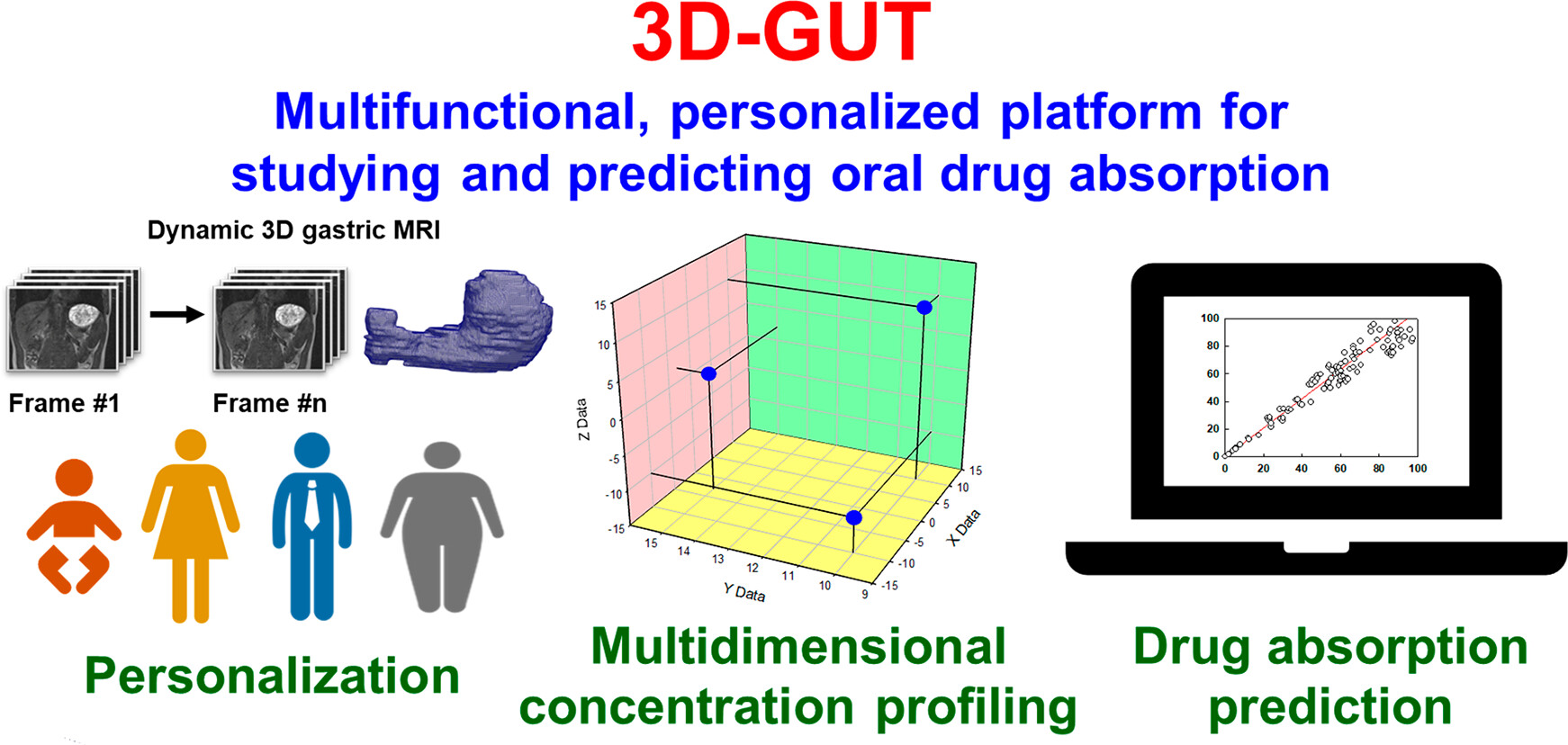
3D virtual gut: a multi-functional, personalized platform for studying and predicting oral drug absorption
Oral drug absorption modelling has been central for physiologically-based pharmacokinetic models (PBPK) which were expected to revolutionize drug development. Instead, PBPK models have delivered unexpectedly large prediction errors and limited functionality, due to their fundamental deficiencies and missing features, including inadequate description of hydrodynamics and missing implementation of protease/nuclease/lipase action, oil phase partitioning, and particle flotation/sedimentation.
This project aims to create a personalized, interactive, 3D virtual model of the human stomach and small intestine that can predict the phase distribution, absorption and food interactions of oral drugs (small molecules, peptides and RNA/DNA), based on advanced time-, space- and phase-resolved in silico profiling of drug concentrations.
The conceptual novelty is the multi-level, multi-scale approach, based on three pillars: (1) development and implementation of new in vitro tools, (2) advanced in silico modelling and (3) new applications of existing in vivo data. Gastrointestinal anatomy and dynamics will be captured via 3D MRI, providing personalization and describing inter-individual variability in organ size and shape. The in vivo parameters will define realistic hydrodynamics, which will be described by computational fluid dynamics and will underpin the mechanistic integration of all main physico-chemical and bio-chemical reactions, at colloidal and molecular levels. New in vitro tools will be developed to study the stability and permeation of peptides and RNA/DNA. Advanced formulation behaviour, drug dissolution, phase distribution and permeation will be integrated via molecular-level models.
This challenging and highly multidisciplinary effort will crystallize in a novel simulation platform of the upper gastrointestinal tract, opening new horizons in personalized medicine, oral drug absorption and food effects research, while facilitating the efforts to abolish animal studies.
Staff





Publications
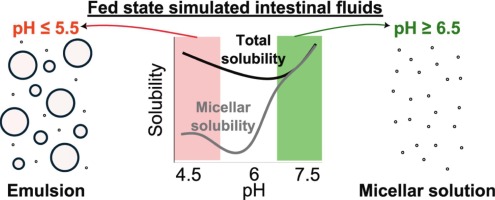
Indirect effects of pH on drug solubility in fed state simulated intestinal fluids
Int. J. Pharm. 2026, 692, 126647
The pH-mediated effect of drug ionization on solubility is well-described. However, pH can also indirectly influence solubility by altering the colloidal structures in human intestinal fluids. This study investigates the indirect pH effect on the apparent solubility of 13 uncharged drugs across a pH range of 4.5 to 7.5 in fed-state simulated intestinal fluids (SIF) composed of taurocholate and lecithin, with or without added lipids (monoolein and/or sodium oleate). A pronounced indirect pH effect on drug solubility was observed when oleate was present in the SIF, whereas monoolein had only a minor effect. Below pH 6.5, sodium oleate was converted to oleic acid, resulting in lipid droplet formation that enhanced lipophilic compound solubility in the total sample (lipid phase + micellar phase), while the micellar solubility remained similar to the reference SIF (without oleate). This resulted in an up to 50-fold increase of the ratio total/micellar drug solubility, which correlated well with drug lipophilicity or its combination with total polar surface area (R2 ≈ 0.8). At higher pH, a lipid phase was not formed because the ionized sodium oleate partitioned in the micellar phase, where it significantly increased drug solubilization. These findings highlight the importance of considering indirect pH effects in solubility assessments by tuning simulated intestinal fluids composition to better reflect in vivo reality.
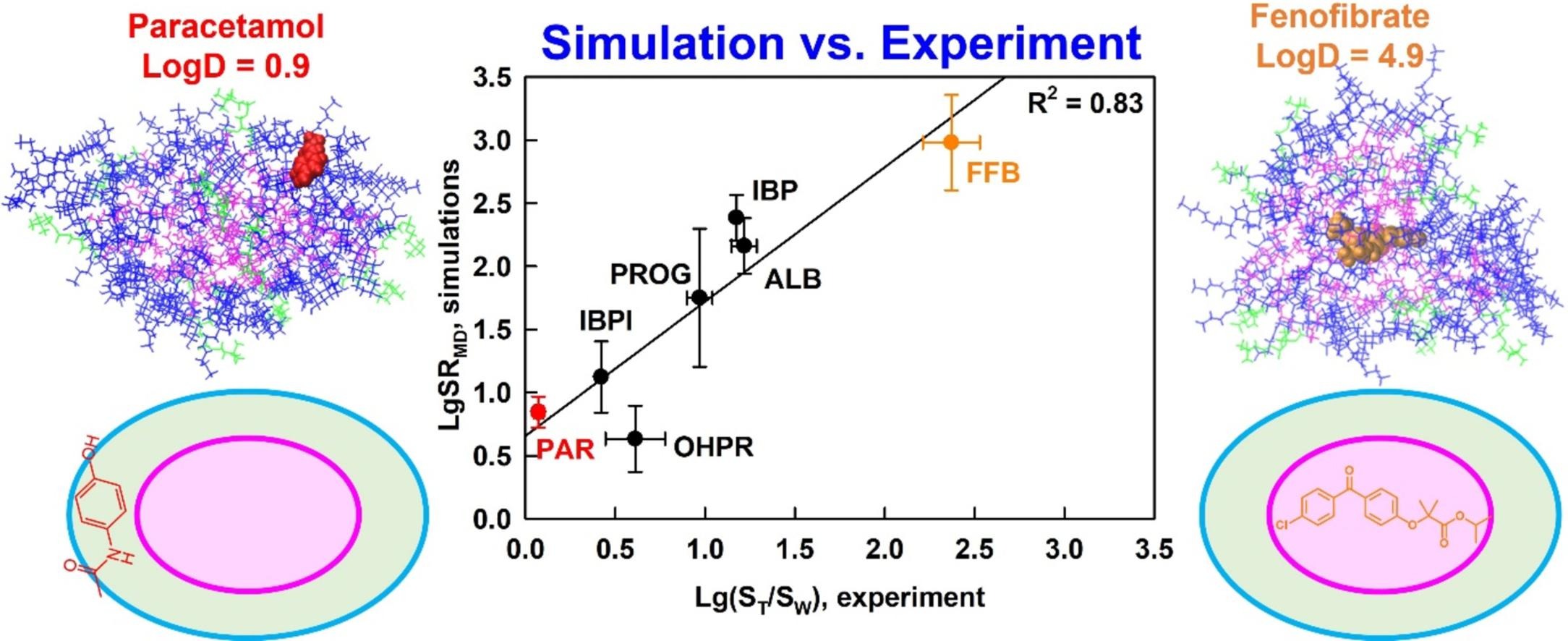
Understanding drug solubilization in intestinal mixed micelles through molecular dynamics simulations
J. Colloid Interface Sci. 2025, 684, 225-234
Hypothesis
Solubilization is a fundamental process that underpins various technologies in the pharmaceutical and chemical industry. However, knowledge of the location, orientation and interactions of solubilized molecules in the micelles is still limited. We expect all-atom molecular dynamics simulations to improve the molecular-level understanding of solubilization and to enable its in silico prediction.
Methods
The solubilization of six drugs in intestinal mixed micelles composed of taurocholate and dioleoyl phosphatidylcholine was simulated by molecular dynamics in explicit water and measured experimentally by liquid chromatography. The location and orientation of the solubilized drugs were visualized by cumulative radial distribution functions and interactions were characterized by radial distribution function ratios and hydrogen bonding.
Findings
A new simulation-derived parameter was defined, which accounts for drug-micelle and drug-water interactions and correlates (R2 = 0.83) with the experimentally measured solubilization. Lipophilicity was found to govern the location of all drugs in the micelle (hydrophobic core, palisade layer or on the surface), while hydrogen bonding was crucial for orientation and solubilization of two of the molecules. The study demonstrates that explicit, hydrogen bond-forming water molecules are vital for accurate prediction of solubilization and provides a comprehensive framework for quantitative studies of drug location and orientation within the micelles.
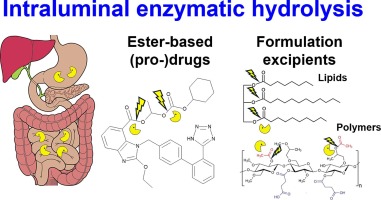
Intraluminal enzymatic hydrolysis of API and lipid or polymeric excipients
Int. J. Pharm. 2025, 675, 125489
The role of intraluminal enzymes for the hydrolysis of active pharmaceutical ingredients (API), prodrugs and pharmaceutical excipients will be reviewed. Carboxylesterases may hydrolyze ester-based API, prodrugs and ester-bond containing polymer excipients, whereas lipases digest lipid formulation excipients, such as mono-, di- and triglycerides. To clarify the conditions that should be mimicked when designing in vitro studies, we briefly review the upper gastrointestinal physiology and provide new data on the inter-individual variability of enzyme activities in human intestinal fluids. Afterwards, the methodology for studying enzymatic hydrolysis of API, prodrugs, lipid and polymeric excipients, as well as the main results that have been obtained, are summarized. In vitro digestion models used to characterize lipid formulations are well described, but data about the hydrolysis of lipid excipients (including surfactants) has been scarce and contradictory. Data on API and prodrug hydrolysis by esterases is available; however, inconsistent use of enzyme types and concentrations limits structure-stability relationships. Hydrolysis of polymer excipients in the lumen has not been significantly explored, with only qualitative data available for cellulose derivates, polyesters, starches, etc. Harmonization of the methodology is required in order to curate larger enzymatic hydrolysis datasets, which will enable mechanistic understanding and theoretical prediction.
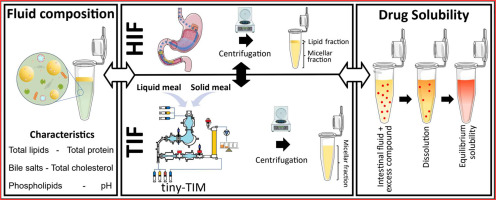
Tiny-TIM intestinal fluids versus human intestinal fluids: A comparison of their composition and solubilizing capacity for poorly soluble drugs
Eur. J. Pharm. Sci. 2025, 211, 107150
The tiny-TIM system offers an in vitro platform for the simulation of physiological processes occurring in human stomach and small intestine aiding in drug product development by predicting the bioperformance of oral formulations under fasted and fed state intake conditions. To assess this in vitro system in terms of its physiological relevance, we performed a detailed analysis of the composition as well as the solubilizing capacity of tiny-TIM intestinal fluids (TIF), and compared this to previously collected and analysed human intestinal fluids (HIF). Moreover, the impact of meal type on TIF composition and solubilising capacity was investigated by using either a liquid meal or a solid meal. In the fasted state, TIF exhibited lower lipid concentrations with a TIF/HIF ratio of 0.27, and elevated bile salt levels (TIF/HIF ratio of 1.8). Fasted state TIF generally overpredicted the solubilizing capacity of HIF, likely due to its higher bile salt concentrations. In the fed state, TIF contained biorelevant lipid concentrations but remained monophasic without phase separation, unlike HIF. This was likely due to higher bile salt levels (5.3 times that of HIF), which solubilized all lipids into the micellar phase. This resulted on average in a 3.3-fold increase in solubility of the poorly water-soluble model compounds in the micellar fraction of TIF as compared to HIF. Shifting from a liquid to a solid meal had minimal impact on TIF composition and solubilizing capacity.
Characterization of human intestinal fluids after the administration of a solid meal
Mol. Pharmaceutics 2025 2025, 22, 12, 7488–7499
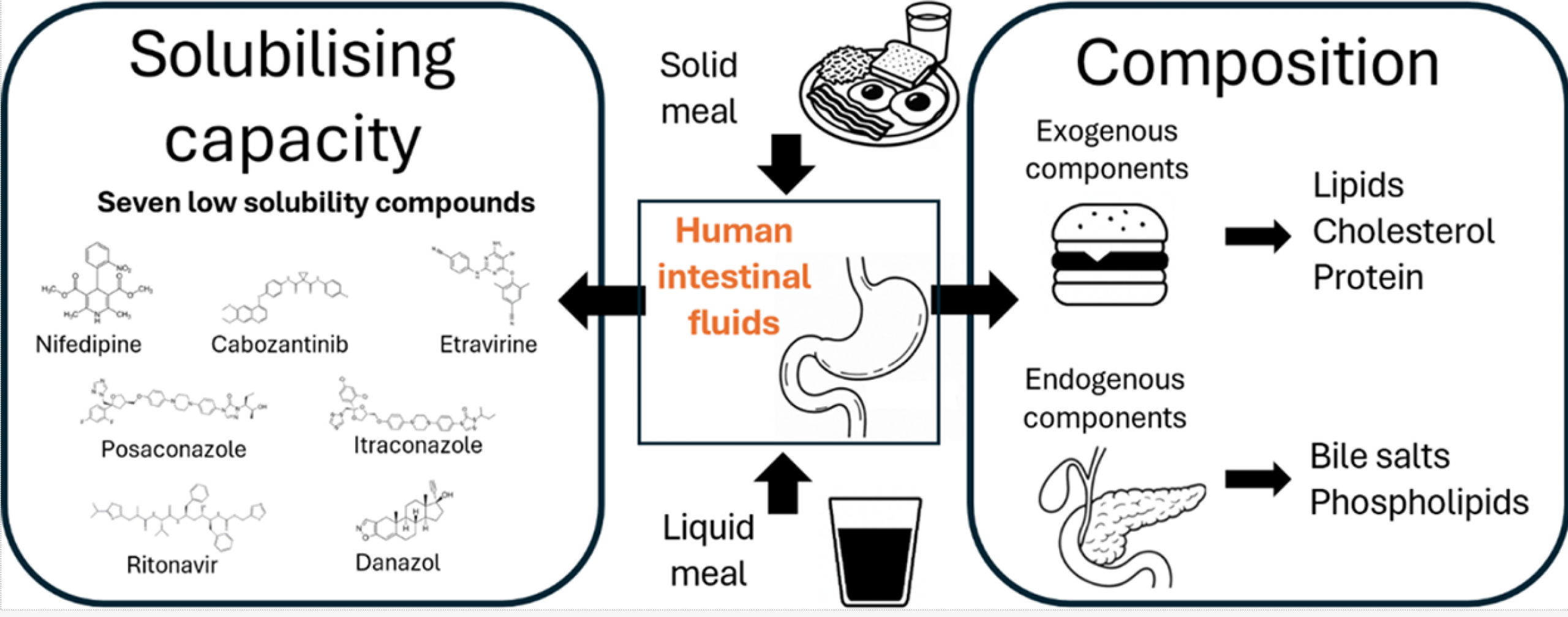
Most available data on the composition and solubilizing properties of postprandial human intestinal fluid (HIF) are derived from studies involving liquid meals. These data inform the development of simulated intestinal fluids, widely used in in vitro assays for predicting intestinal drug behavior. However, the typical human diet primarily consists of solid meals, and the physical form of food has been shown to influence gastrointestinal transit and digestion, thereby affecting drug disposition and bioavailability. This study compares the characteristics of fed-state HIF collected after solid meal ingestion (SM-HIF) with previously published data on pooled liquid meal-derived HIF (LM-HIF) and newly generated data from individual LM-HIF samples. Time-dependent samples were analyzed over 180- and 90 min postprandial sampling periods to assess compositional changes following the administration of a solid and liquid meal, respectively. In addition, pooled samples were used to evaluate the solubilizing capacity for seven lipophilic model compounds. After intake of the solid meal, duodenal concentrations of exogenous (lipids, cholesterol, proteins) and endogenous (bile salts, phospholipids) components gradually increased to peak levels reached after 45−75 min. After 180 min, lipid and protein concentrations were still elevated compared to fasted state levels. In comparison to the liquid meal, the ingestion of the solid meal resulted in reduced concentrations of exogenous components, while endogenous components (bile salts and phospholipids) were relatively similar. For most compounds, the reduction in lipid content led to diminished solubilizing capacity of SM-HIF compared to LM-HIF when considering the combined micellar and lipid fractions. In contrast, the solubilizing capacity of the micellar fraction as such was largely independent of the meal type. Both the composition (particularly the micellar lipid concentration) and the solubilizing capacity of SM-HIF were highly variable between pools, albeit to a lesser extent than in LMHIF. The findings of this study highlight that the physical form of the meal influences the composition and solubilizing capacity of HIF. These insights should be taken into account when refining biorelevant media for in vitro models to better predict food effects during drug product development.
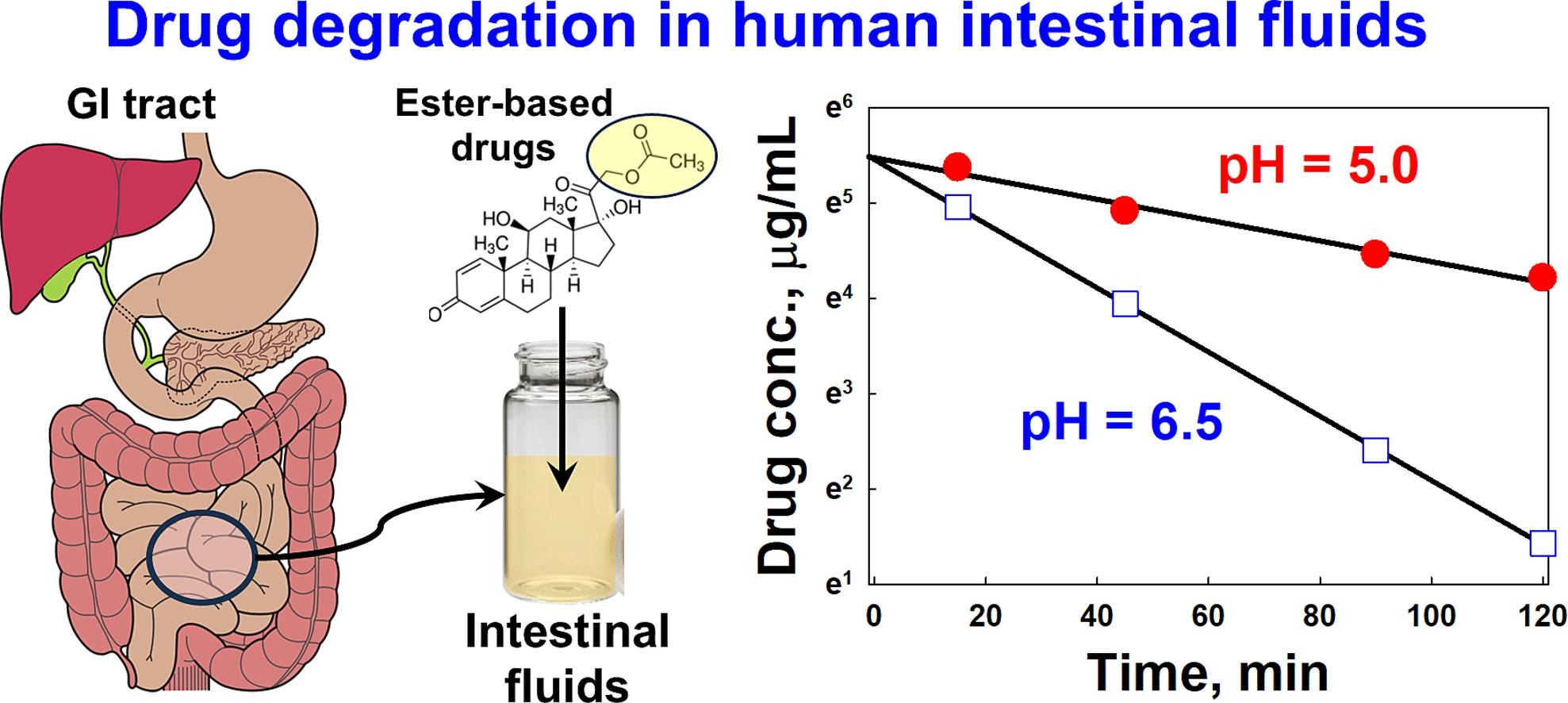
Enzymatic prodrug degradation in the fasted and fed small intestine: In vitro studies and interindividual variability in human aspirates
Int. J. Pharm. 2024, 649, 123654
The aim of the current study was (1) to develop an automation-based protocol for in vitro assessment of enzymatic drug stability at fasted- and fed-state intestinal conditions, (2) to characterize the inter-individual variability of drug degradation in fasted- and fed-state human intestinal fluids, and (3) to compare the obtained in vitro results to drug degradation in human intestinal fluids by taking variability into account. In human intestinal fluids, drug degradation displayed large inter-individual variability, with coefficients of variance generally ranging between 30 and 70 %. The effect of food on the inter-individual variability was highly dependent on the type of drug. The increase of pH in the range between 5.0 and 7.0 significantly accelerated the degradation rate of the studied drugs both in the in vitro and ex vivo experiments. In contrast, the increase of bile salt and phospholipid concentrations in the in vitro screen decreased strongly the degradation rate of the hydrophobic drugs. The developed automated in vitro screen mimicked relatively well the ex vivo degradation of all drugs in the fasted state, whereas in the fed state the degradation of only one of the drugs was adequately reproduced.
Mechanisms of dissolution and crystallization of amorphous glibenclamide
Int. J. Pharm. 2024, 666, 124820
Amorphous solid dispersions enhance the dissolution and oral bioavailability of poorly water-soluble drugs. However, the link between polymer properties and formulation performance has not been fully clarified yet. We studied the effect of hydroxypropyl cellulose (HPC) polymers molecular weight (Mw) on the storage stability, dissolution kinetics and supersaturation stability of spray-dried amorphous glibenclamide (GLB) formulations. The solid-state stability of amorphous GLB during storage was significantly enhanced by both the 40 kDa (HPC-SSL) and 84 kDa (HPC-L) polymers, regardless of Mw differences. In contrast, HPC-SSL maintained significantly higher aqueous drug concentrations during dissolution, compared to HPC-L (its higher Mw analogue). Dedicated dissolution experiments, in situ optical microscopy and solid-state characterization revealed that aqueous drug concentrations were determined by the interplay between crystallization inhibition, drug ionization, wetting and solubilization effects: (1) HPC prevents surface nucleation, hence inhibiting crystallization, (2) intestinal colloids (bile salts and phospholipids) increase supersaturated drug concentrations via wetting and solubilization effects and (3) pH and drug ionization severely impact the degree of supersaturation. The better performance of the lower Mw HPC-SSL was due to its superior inhibition of surface crystallization during dissolution. These insights into the molecular mechanisms of dissolution and crystallization of amorphous solids provide foundation for rational formulation development.

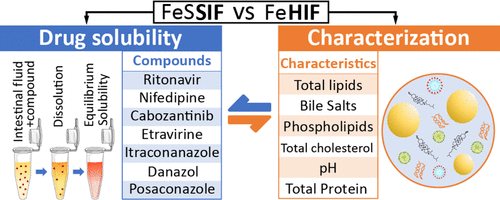
Understanding the impact of lipids on the solubilizing capacity of human intestinal fluids
Mol. Pharm. 2024, 21, 6398-6410
Lipids in human intestinal fluids (HIF) form various structures, resulting in phase separation in the form of a lipid fraction and a micellar aqueous fraction. Currently used fed state simulated intestinal fluids (SIF) lack phase separation, highlighting the need for a deeper understanding of the effect of these fractions on intestinal drug solubilization in HIF to improve simulation accuracy. In this study, duodenal fluids aspirated from 21 healthy volunteers in fasted, early fed, and late fed states were used to generate 7 HIF pools for each prandial state. The apparent solubility of seven lipophilic model drugs was measured across these HIF pools, differentiating between the micellar fraction and the total sample (including both micellar and lipid fractions). The solubilizing capacities of these fluids were analyzed in relation to their composition, including total lipids, bile salts, phospholipids, total cholesterol, pH, and total protein. The solubility data generated in this work demonstrated that current fed state SIF effectively predicted the average solubility in the micellar fraction of HIF but failed to discern the considerable variability between HIF pools. Furthermore, the inclusion of a lipid fraction significantly enhanced the solubility of fed state HIF pools, resulting on average in a 13.9-fold increase in solubilizing capacity across the seven model compounds. Although the average composition of the fluids was consistent with previous studies, substantial variability was observed in micellar lipid concentrations, despite relatively stable total lipid concentrations. This variability is critical, as evidenced by the strong correlations between the solubilizing capacity of the micellar fraction and its micellar lipid concentrations. Additionally, this study identified that fluctuations in bile salt concentrations and pH contributed to the observed variability in micellar lipid concentration. In summary, the influence of the lipid fraction on solubility was 2-fold: it enhanced the solubility of lipophilic drugs in the total fluid, and contributed to the variability in the solubilizing capacity of the micellar fraction.
Assessment of food effects during clinical development
Int. J. Pharm. 2023, 635, 122758
Food-drug interactions frequently hamper oral drug development due to various physicochemical, physiological and formulation-dependent mechanisms. This has stimulated the development of a range of promising biopharmaceutical assessment tools which, however, lack standardized settings and protocols. Hence, this manuscript aims to provide an overview of the general approach and the methodology used in food effect assessment and prediction. For in vitro dissolution-based predictions, the expected food effect mechanism should be carefully considered when selecting the level of complexity of the model, together with its drawbacks and advantages. Typically, in vitro dissolution profiles are then incorporated into physiologically based pharmacokinetic models, which can estimate the impact of food-drug interactions on bioavailability within 2-fold prediction error, at least. Positive food effects related to drug solubilization in the GI tract are easier to predict than negative food effects. Preclinical animal models also provide a good level of food effect prediction, with beagle dogs remaining the gold standard. When solubility-related food-drug interactions have large clinical impact, advanced formulation approaches can be used to improve fasted state pharmacokinetics, hence decreasing the fasted/fed difference in oral bioavailability. Finally, the knowledge from all studies should be combined to secure regulatory approval of the labelling instructions.
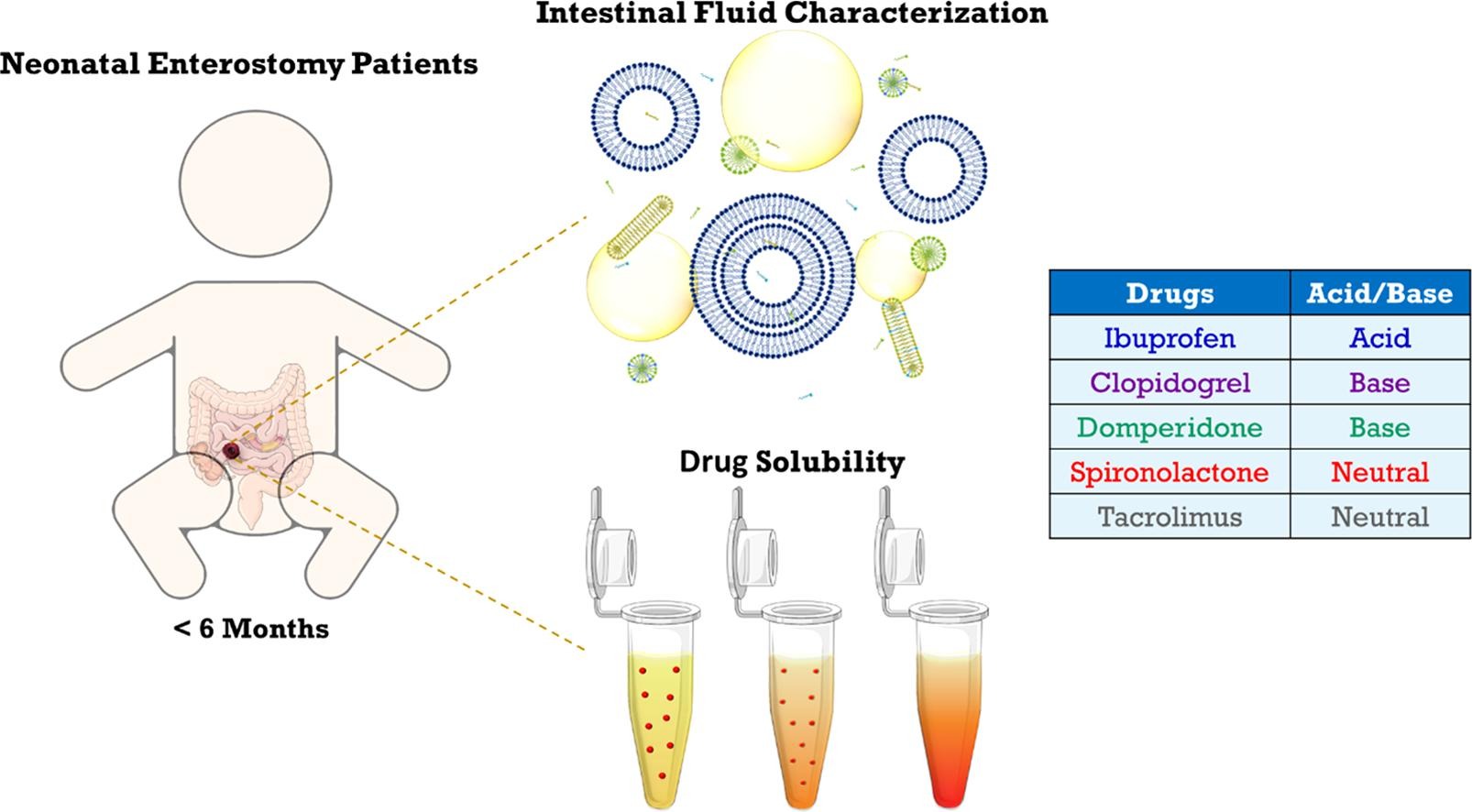
Characterization of neonatal and infant enterostomy fluids – Part II: Drug solubility
Int. J. Pharm. 2023, 642, 123141
Previous research revealed marked differences in the composition of intestinal fluids between infants and adults. To explore the impact on the solubilization of orally administered drugs, the present study assessed the solubility of five poorly water-soluble, lipophilic drugs in intestinal fluid pools from 19 infant enterostomy patients (infant HIF). For some but not all drugs, the average solubilizing capacity of infant HIF was similar to that of HIF obtained from adults (adult HIF) in fed conditions. Commonly used fed state simulated intestinal fluids (FeSSIF(-V2)) predicted fairly well drug solubility in the aqueous fraction of infant HIF, but did not account for the substantial solubilization by the lipid phase of infant HIF. Despite similarities in the average solubilities of some drugs in infant HIF and adult HIF or SIF, the underlying solubilization mechanisms likely differ, considering important compositional differences (e.g., low bile salt levels). Finally, the huge variability in composition of infant HIF pools resulted in a highly variable solubilizing capacity, potentially causing variations in drug bioavailability. The current study warrants future research focusing on (i) understanding the mechanisms underlying drug solubilization in infant HIF and (ii) evaluating the sensitivity of oral drug products to interpatient variations in drug solubilization.
Voices in molecular pharmaceutics: Meet Dr. Zahari Vinarov, who unites physical chemistry and pharmacy to tackle fundamental and industrial biopharmaceutical challenges
Mol. Pharm. 2023, 20, 5949 - 5951
I am currently an associate professor, leading several research projects funded by the pharmaceutical industry or public research agencies. One of these projects is a five-year (2022–2027) national grant (the 3D GUT project), which aims to create a personalized, interactive, 3D virtual model of the human stomach and small intestine that can predict the phase distribution, absorption, and food interactions of oral drugs (small molecules, peptides, and RNA/DNA), based on advanced time-, space-, and phase-resolved in silico profiling of drug concentrations (Figure 1). To achieve this ambitious goal, our team will (1) capture gastrointestinal anatomy and dynamics via 3D MRI, providing personalization and describing interindividual variability in organ size and shape, (2) use computational fluid dynamics to study the real hydrodynamics at anatomically and physiologically relevant conditions, and (3) integrate all main physicochemical and biochemical reactions, at colloidal and molecular levels, including advanced formulation behavior, drug dissolution, phase distribution, and permeation. This challenging and highly multidisciplinary effort aims to lead to a novel simulation platform of the upper gastrointestinal tract, opening new horizons in personalized medicine, oral drug absorption, and food effects research, while facilitating the efforts to abolish animal studies.