Assoc. Prof. Zahari P. Vinarov, Ph.D. Chemistry, Ph.D Pharmacy
Head of the Department
Interests
- Oral formulation development and biopharmaceutical characterization
- Intestinal absorption mechanisms
- Amorphous solid dispersions and lipid formulations
- In vitro modelling of the gastrointestinal tract
- Physicochemical aspects of formulation and biopharmaceutics
Bio
Publications
Featured publications
Mechanisms of dissolution and crystallization of amorphous glibenclamide
Amorphous solid dispersions enhance the dissolution and oral bioavailability of poorly water-soluble drugs. However, the link between polymer properties and formulation performance has not been fully clarified yet. We studied the effect of hydroxypropyl cellulose (HPC) polymers molecular weight (Mw) on the storage stability, dissolution kinetics and supersaturation stability of spray-dried amorphous glibenclamide (GLB) formulations. The solid-state stability of amorphous GLB during storage was significantly enhanced by both the 40 kDa (HPC-SSL) and 84 kDa (HPC-L) polymers, regardless of Mw differences. In contrast, HPC-SSL maintained significantly higher aqueous drug concentrations during dissolution, compared to HPC-L (its higher Mw analogue). Dedicated dissolution experiments, in situ optical microscopy and solid-state characterization revealed that aqueous drug concentrations were determined by the interplay between crystallization inhibition, drug ionization, wetting and solubilization effects: (1) HPC prevents surface nucleation, hence inhibiting crystallization, (2) intestinal colloids (bile salts and phospholipids) increase supersaturated drug concentrations via wetting and solubilization effects and (3) pH and drug ionization severely impact the degree of supersaturation. The better performance of the lower Mw HPC-SSL was due to its superior inhibition of surface crystallization during dissolution. These insights into the molecular mechanisms of dissolution and crystallization of amorphous solids provide foundation for rational formulation development.

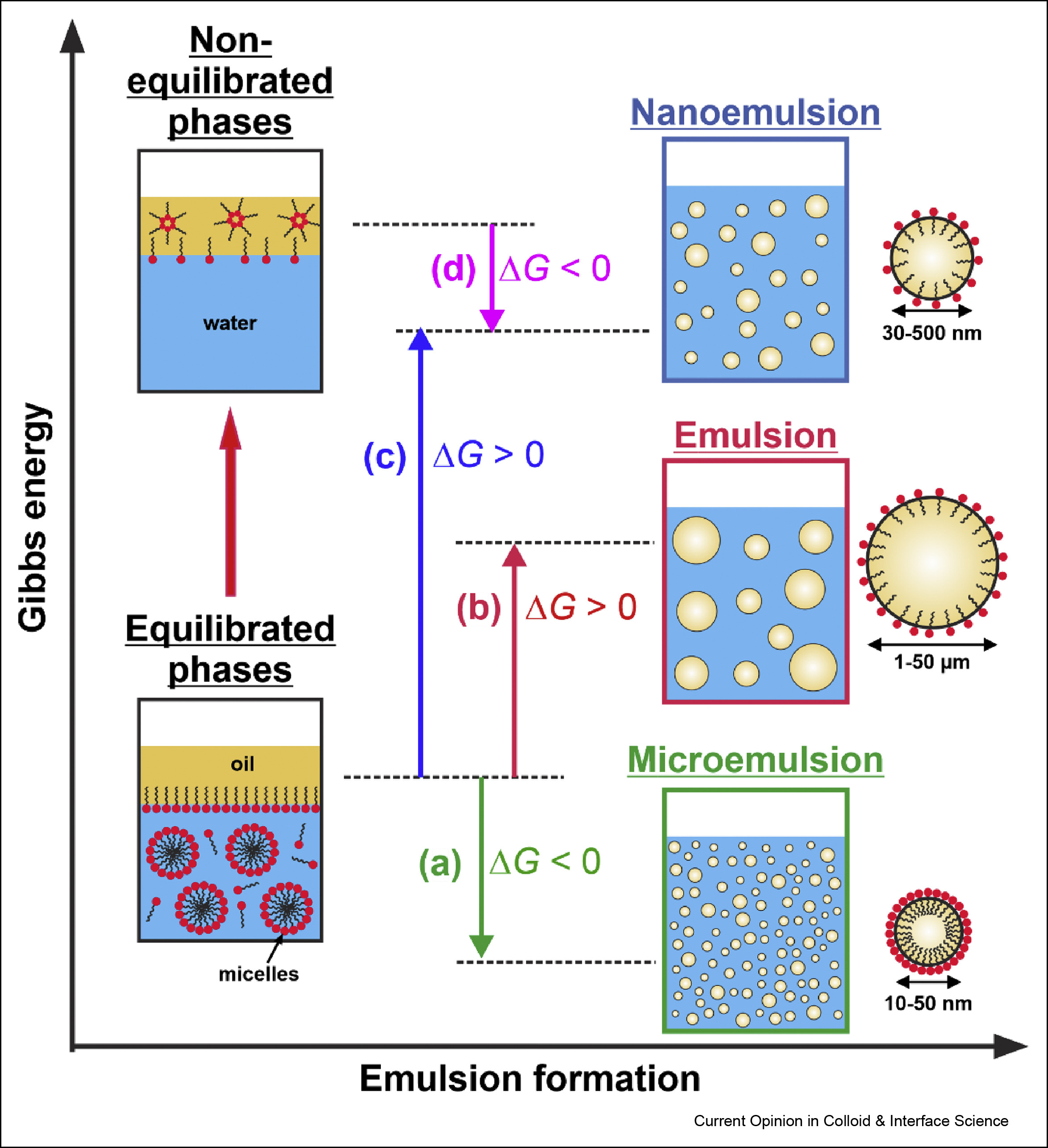
Self-emulsification in chemical and pharmaceutical technologies
The interest in the low energy self-emulsification techniques has exploded in the recent years, driven by three main trends: by the transition to “greener” technologies in both its aspects—less energy consumption and replacement of the petrochemicals by natural ingredients; by the costly and maintenance demanding equipment for nanoemulsification; and by the quest for efficient and robust self-emulsifying formulations for oral drug delivery. Here, we first present a brief overview of the main known low-energy methods for nanoemulsion formation, focusing on their mechanistic understanding and discussing some recent advances in their development and applications. Next, we review three conceptually new approaches for self-emulsification in chemical technologies, discovered in the last several years. The colloidal features and the specific requirements of the self-emulsifying drug-delivery systems (SEDDS) are also discussed briefly. Finally, we summarize the current trends and the main challenges in this vivid research area.
Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review
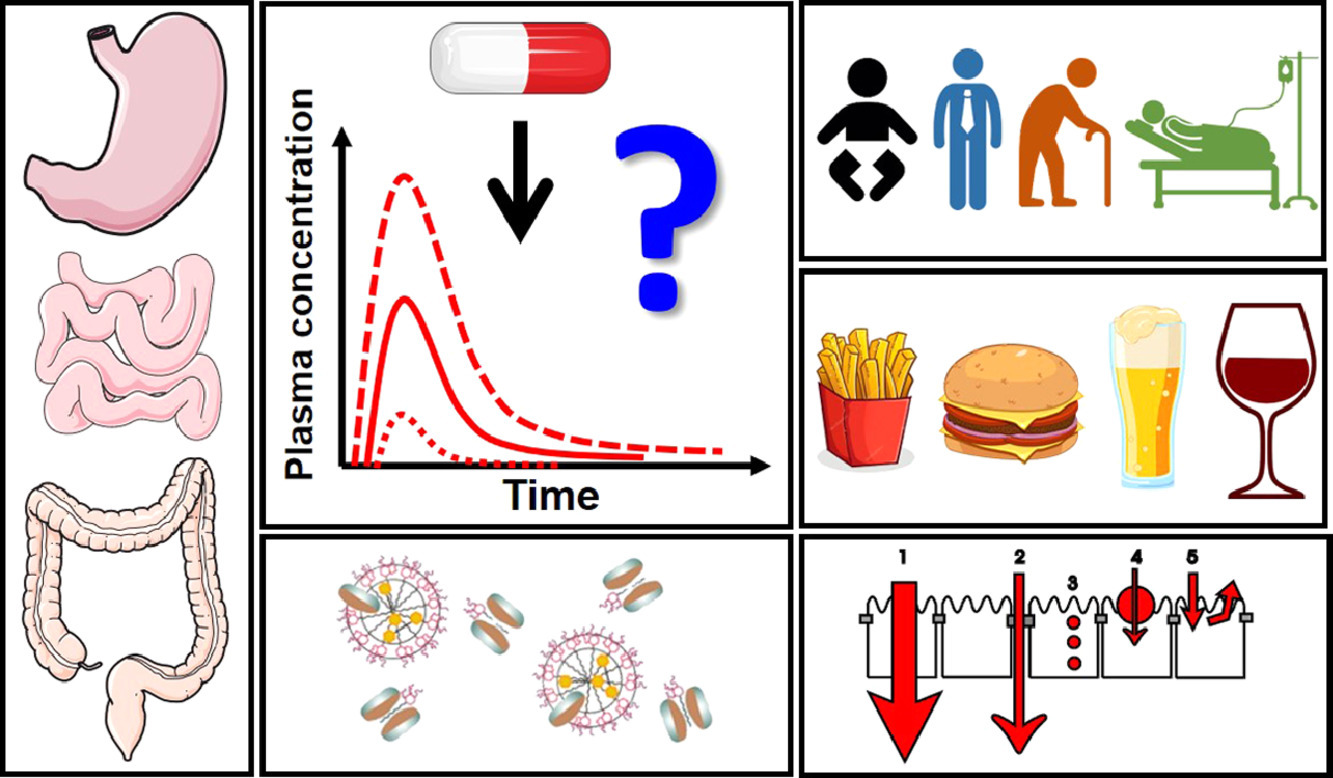
The absorption of oral drugs is frequently plagued by significant variability with potentially serious therapeutic consequences. The source of variability can be traced back to interindividual variability in physiology, differences in special populations (age- and disease-dependent), drug and formulation properties, or food-drug interactions. Clinical evidence for the impact of some of these factors on drug pharmacokinetic variability is mounting: e.g. gastric pH and emptying time, small intestinal fluid properties, differences in pediatrics and the elderly, and surgical changes in gastrointestinal anatomy. However, the link of colonic factors variability (transit time, fluid composition, microbiome), sex differences (male vs. female) and gut-related diseases (chronic constipation, anorexia and cachexia) to drug absorption variability has not been firmly established yet. At the same time, a way to decrease oral drug pharmacokinetic variability is provided by the pharmaceutical industry: clinical evidence suggests that formulation approaches employed during drug development can decrease the variability in oral exposure. This review outlines the main drivers of oral drug exposure variability and potential approaches to overcome them, while highlighting existing knowledge gaps and guiding future studies in this area.
Current challenges and future perspectives in oral absorption research: An opinion of the UNGAP network
Although oral drug delivery is the preferred administration route and has been used for centuries, modern drug discovery and development pipelines challenge conventional formulation approaches and highlight the insufficient mechanistic understanding of processes critical to oral drug absorption. This review presents the opinion of UNGAP scientists on four key themes across the oral absorption landscape: (1) specific patient populations, (2) regional differences in the gastrointestinal tract, (3) advanced formulations and (4) food-drug interactions. The differences of oral absorption in pediatric and geriatric populations, the specific issues in colonic absorption, the formulation approaches for poorly water-soluble (small molecules) and poorly permeable (peptides, RNA etc.) drugs, as well as the vast realm of food effects, are some of the topics discussed in detail. The identified controversies and gaps in the current understanding of gastrointestinal absorption-related processes are used to create a roadmap for the future of oral drug absorption research.

Mechanisms of drug solubilization by polar lipids in biorelevant media
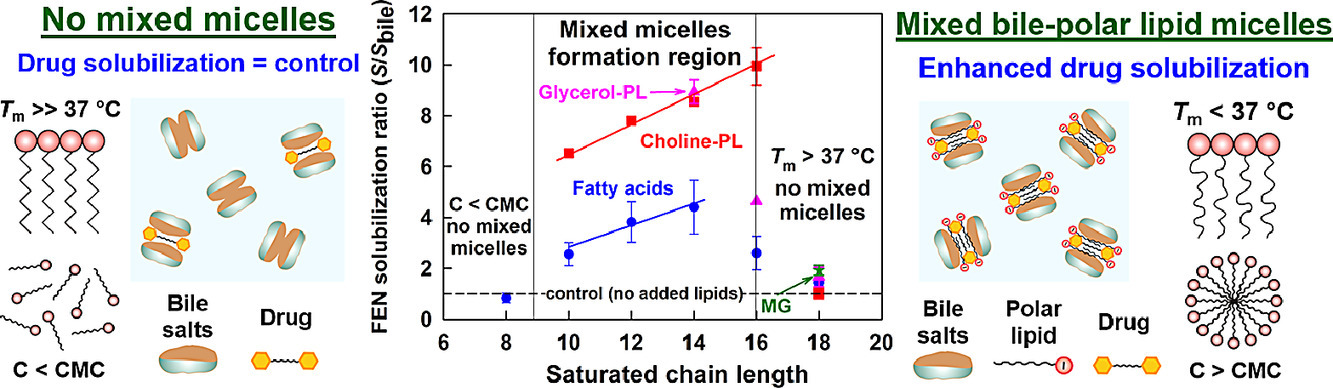
Despite the widespread use of lipid excipients in both academic research and oral formulation development, rational selection guidelines are still missing. In the current study, we aimed to establish a link between the molecular structure of commonly used polar lipids and drug solubilization in biorelevant media. The solubilization of fenofibrate by 13 phospholipids, 11 fatty acids and 2 monoglycerides was studied by an in vitro model of the upper GI tract. The main trends were verified with progesterone and danazol. It was revealed that to alter drug solubilization in biorelevant media, the polar lipids must form mixed colloidal aggregates with the bile. Such aggregates are formed when: (1) the polar lipid is used at a sufficiently high concentration (relative to its mixed critical micellar concentration) and (2) its hydrophobic chain has a melting temperature (Tm) < 37 °C. When these two conditions are met, the increased polar lipid chain length increases the drug solubilization capacity. Hence, long chain (C18) unsaturated polar lipids show best drug solubilization, due to the combination of long chain length and low Tm. Polar lipids with Tm significantly higher than 37 °C (e.g. C16 and C18 saturated compounds) do not impact drug solubilization in biorelevant media, due to limited association in mixed colloidal aggregates. The hydrophilic head group also has a dramatic impact on the drug solubilization enhancement, with polar lipids performance decreasing in the order [choline phospholipids] > [monoglycerides] > [fatty acids]. As both the acyl chain and head group types are structural features of the polar lipids, and not of the solubilized drugs, the described trends in drug solubilization should hold true for a variety of hydrophobic molecules.
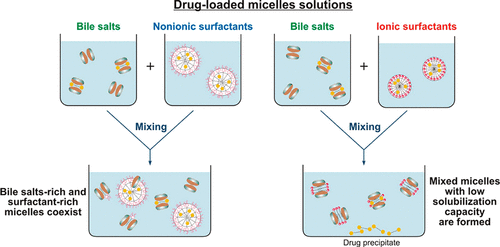
Effect of surfactant-bile interactions on the solubility of hydrophobic drugs in biorelevant dissolution media
Biorelevant dissolution media (BDM) methods are commonly employed to investigate the oral absorption of poorly water-soluble drugs. Despite the significant progress in this area, the effect of commonly employed pharmaceutical excipients, such as surfactants, on the solubility of drugs in BDM has not been characterized in detail. The aim of this study is to clarify the impact of surfactant-bile interactions on drug solubility by using a set of 12 surfactants, 3 model hydrophobic drugs (fenofibrate, danazol, and progesterone) and two types of BDM (porcine bile extract and sodium taurodeoxycholate). Drug precipitation and sharp nonlinear decrease in the solubility of all studied drugs is observed when drug-loaded ionic surfactant micelles are introduced in solutions of both BDM, whereas the drugs remain solubilized in the mixtures of nonionic polysorbate surfactants + BDM. One-dimensional and diffusion-ordered 1 H NMR spectroscopy show that mixed bile salt + surfactant micelles with low drug solubilization capacity are formed for the ionic surfactants. On the other hand, separate surfactant-rich and bile salt-rich micelles coexist in the nonionic polysorbate surfactant + bile salt mixtures, explaining the better drug solubility in these systems. The nonionic alcohol ethoxylate surfactants show intermediate behavior. The large dependence of the drug solubility on surfactant-bile interactions (in which the drug molecules do not play a major role per se) highlights how the complex interplay between excipients and bile salts can significantly change one of the key parameters which governs the oral absorption of poorly water-soluble drugs, viz. the drug solubility in the intestinal fluids.
Most recent publications
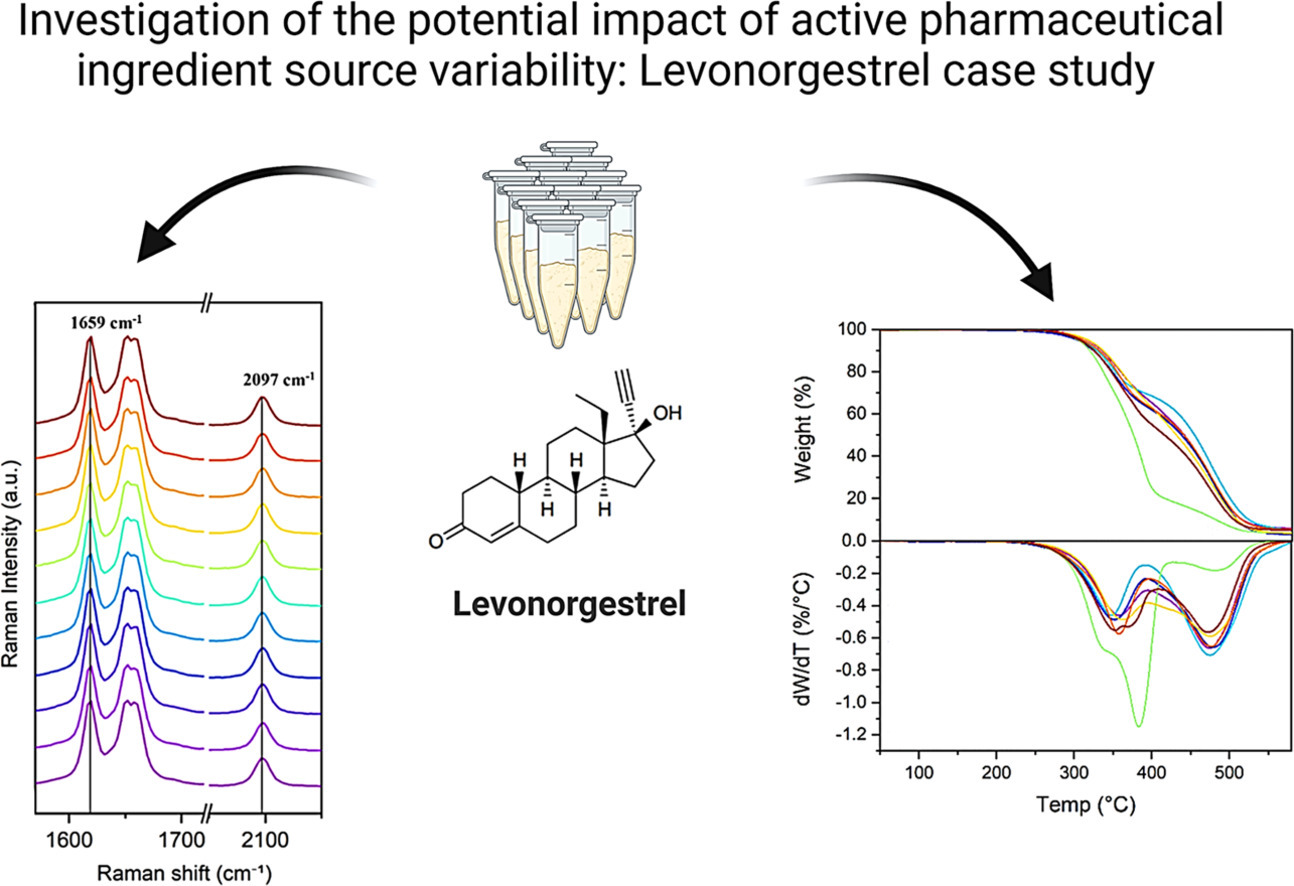
Preformulation studies of levonorgestrel: A supplier variation analysis
Levonorgestrel (LNG) is a second-generation synthetic progestogen that has been widely used in oral tablets for emergency contraception, intrauterine devices and transdermal implants. The main objectives of the present work were to evaluate LNG drug substance variability with respect to polymorphism and purity, and to investigate the stability of the compound in the selected organic solvents, pharmaceutical vehicles and biorelevant media. Solid-state analysis of LNG samples from eleven different suppliers were conducted by different analytical techniques, including Fourier transform infrared (FTIR), Raman, solid-state and liquid-state nuclear magnetic resonance (NMR) spectroscopies, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), powder X-ray diffraction (XRPD), wide-angle X-ray scattering (WAXS), small-angle X-ray scattering (SAXS) and scanning electron microscopy (SEM). In addition, dynamic vapour sorption (DVS) was performed to investigate the hygroscopicity of LNG. The comprehensive solid-state investigation of LNG from different sources showed only minor variations related to their thermal properties. Overall, all suppliers delivered the same polymorphic form of LNG and the compound displayed high physical stability under elevated temperatures and across relevant organic solvents, suggesting limited risks associated with polymorphic changes during processing. Moreover, the equilibrium solubility was obtained in a wide range of relevant organic solvents, dissolution media and pharmaceutical vehicles, which would support development of new dosage forms containing LNG.
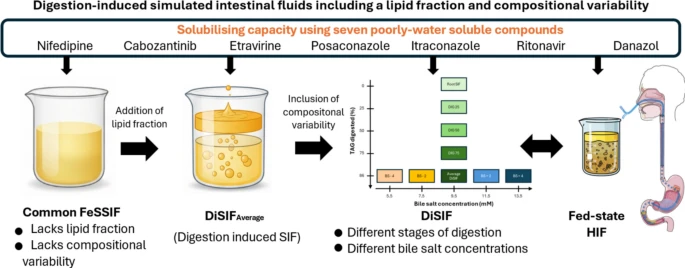
Exploring digestion-induced fed-state simulated intestinal media with a lipid fraction and compositional variability
Food effects on intestinal drug solubility and dissolution are a critical consideration in drug development, commonly investigated using simulated intestinal fluids (SIF) such as FaSSIF and FeSSIF. While these media represent average fasted- and fed-state conditions, their lack of a lipid fraction and disregard for compositional variability limit their physiological relevance and predictive power. To address these limitations, this proof-of-concept study introduces a novel set of fed-state SIF, termed digestion-induced SIF (DiSIF), featuring two key enhancements: (i) inclusion of a physiologically relevant lipid fraction generated through in vitro digestion of a liquid meal, and (ii) incorporation of variability via modulation of bile salt concentration and stage of digestion. Nine DiSIF media were developed to reflect both the average composition and the variability observed in fed-state human intestinal fluids (HIF). Overall, DiSIF media adequately predicted the micellar solubility of seven poorly water-soluble model compounds. Unlike commonly used SIF, they also enabled estimation of drug solubilization in total samples containing both micellar and lipid fractions, as observed in fed-state HIF. Furthermore, the media’s compositional variability allowed for the prediction of compound sensitivity to physiological variability with relative accuracy. These findings support further refinement and validation of DiSIF media as a versatile tool for formulation development and food-effect prediction in oral drug delivery.
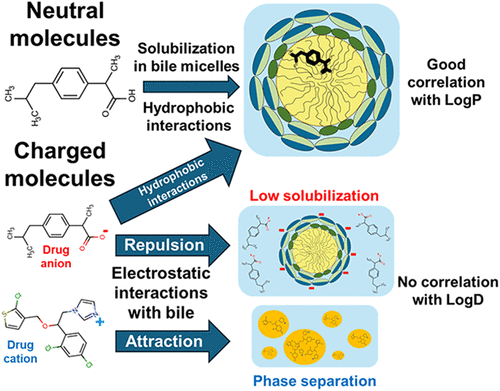
Drug solubilization in simulated intestinal fluids vs lipophilicity: Does charge matter?
Solubilization of poorly water-soluble drugs in human intestinal fluids influences oral absorption and is linked to food effects. Current empirical equations for calculating intestinal solubilization via lipophilicity are built on limited data and do not adequately account for drug ionization. We aim to expand the data set and build a model to clarify the link between lipophilicity and solubilization for charged compounds. We determined the aqueous solubility, octanol–water partition coefficient, and solubilization in fed-state simulated intestinal fluids (FeSSIF) of 26 hydrophobic drugs. Combined with literature data, a good correlation (R2 = 0.74, n = 198) between intestinal solubilization and LogP/D was observed. However, data segregation showed that the solubilization of neutral compounds correlated very well with LogP (R2 = 0.89, n = 114), whereas the correlation with LogD was lost for the charged compounds (R2 = 0.40, n = 84). To better understand this behavior, the pH of FeSSIF was varied to study the solubilization of the same compounds in the neutral and charged states. While a very good correlation between solubilization and LogD was observed in the neutral state of the compounds (R2 = 0.92, n = 8), the correlation was again lost (R2 = 0.02, n = 4) in their charged state. Electrostatic interactions were suggested to play a key role in the unexpectedly low solubilization of anionic drugs and in the phase separation observed for cationic drugs. The presented insights further advance the understanding of the solubilization of hydrophobic drugs in biorelevant media and provide a foundation for broader and improved modeling approaches.
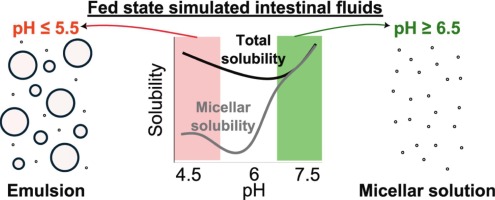
Indirect effects of pH on drug solubility in fed state simulated intestinal fluids
The pH-mediated effect of drug ionization on solubility is well-described. However, pH can also indirectly influence solubility by altering the colloidal structures in human intestinal fluids. This study investigates the indirect pH effect on the apparent solubility of 13 uncharged drugs across a pH range of 4.5 to 7.5 in fed-state simulated intestinal fluids (SIF) composed of taurocholate and lecithin, with or without added lipids (monoolein and/or sodium oleate). A pronounced indirect pH effect on drug solubility was observed when oleate was present in the SIF, whereas monoolein had only a minor effect. Below pH 6.5, sodium oleate was converted to oleic acid, resulting in lipid droplet formation that enhanced lipophilic compound solubility in the total sample (lipid phase + micellar phase), while the micellar solubility remained similar to the reference SIF (without oleate). This resulted in an up to 50-fold increase of the ratio total/micellar drug solubility, which correlated well with drug lipophilicity or its combination with total polar surface area (R2 ≈ 0.8). At higher pH, a lipid phase was not formed because the ionized sodium oleate partitioned in the micellar phase, where it significantly increased drug solubilization. These findings highlight the importance of considering indirect pH effects in solubility assessments by tuning simulated intestinal fluids composition to better reflect in vivo reality.
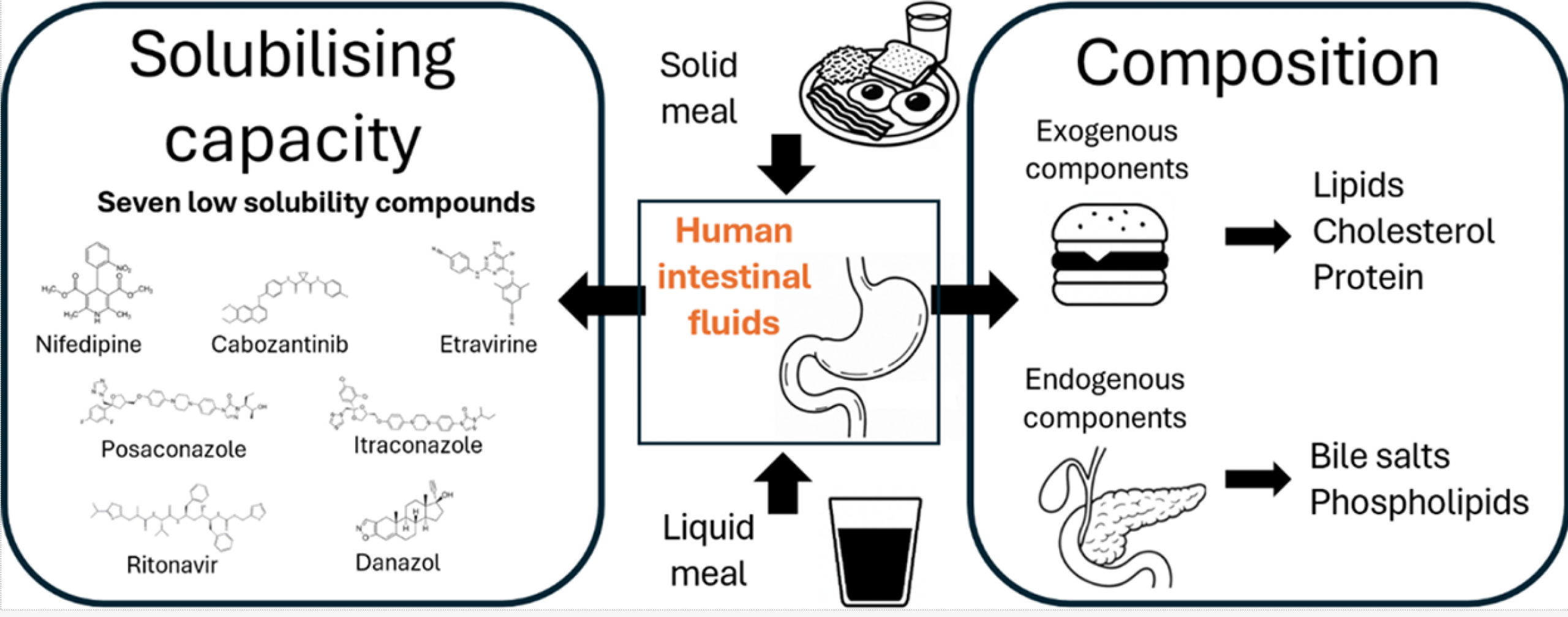
Characterization of human intestinal fluids after the administration of a solid meal
Most available data on the composition and solubilizing properties of postprandial human intestinal fluid (HIF) are derived from studies involving liquid meals. These data inform the development of simulated intestinal fluids, widely used in in vitro assays for predicting intestinal drug behavior. However, the typical human diet primarily consists of solid meals, and the physical form of food has been shown to influence gastrointestinal transit and digestion, thereby affecting drug disposition and bioavailability. This study compares the characteristics of fed-state HIF collected after solid meal ingestion (SM-HIF) with previously published data on pooled liquid meal-derived HIF (LM-HIF) and newly generated data from individual LM-HIF samples. Time-dependent samples were analyzed over 180- and 90 min postprandial sampling periods to assess compositional changes following the administration of a solid and liquid meal, respectively. In addition, pooled samples were used to evaluate the solubilizing capacity for seven lipophilic model compounds. After intake of the solid meal, duodenal concentrations of exogenous (lipids, cholesterol, proteins) and endogenous (bile salts, phospholipids) components gradually increased to peak levels reached after 45−75 min. After 180 min, lipid and protein concentrations were still elevated compared to fasted state levels. In comparison to the liquid meal, the ingestion of the solid meal resulted in reduced concentrations of exogenous components, while endogenous components (bile salts and phospholipids) were relatively similar. For most compounds, the reduction in lipid content led to diminished solubilizing capacity of SM-HIF compared to LM-HIF when considering the combined micellar and lipid fractions. In contrast, the solubilizing capacity of the micellar fraction as such was largely independent of the meal type. Both the composition (particularly the micellar lipid concentration) and the solubilizing capacity of SM-HIF were highly variable between pools, albeit to a lesser extent than in LMHIF. The findings of this study highlight that the physical form of the meal influences the composition and solubilizing capacity of HIF. These insights should be taken into account when refining biorelevant media for in vitro models to better predict food effects during drug product development.