Sonya R. Tsibranska-Gyoreva, Ph.D.
Interests
- Emulsions and Emulsification
- Nanoparticle Synthesis
- Bulk Rheology of Emulsions
- Molecular Dynamic Simulations
Publications
Most recent publications
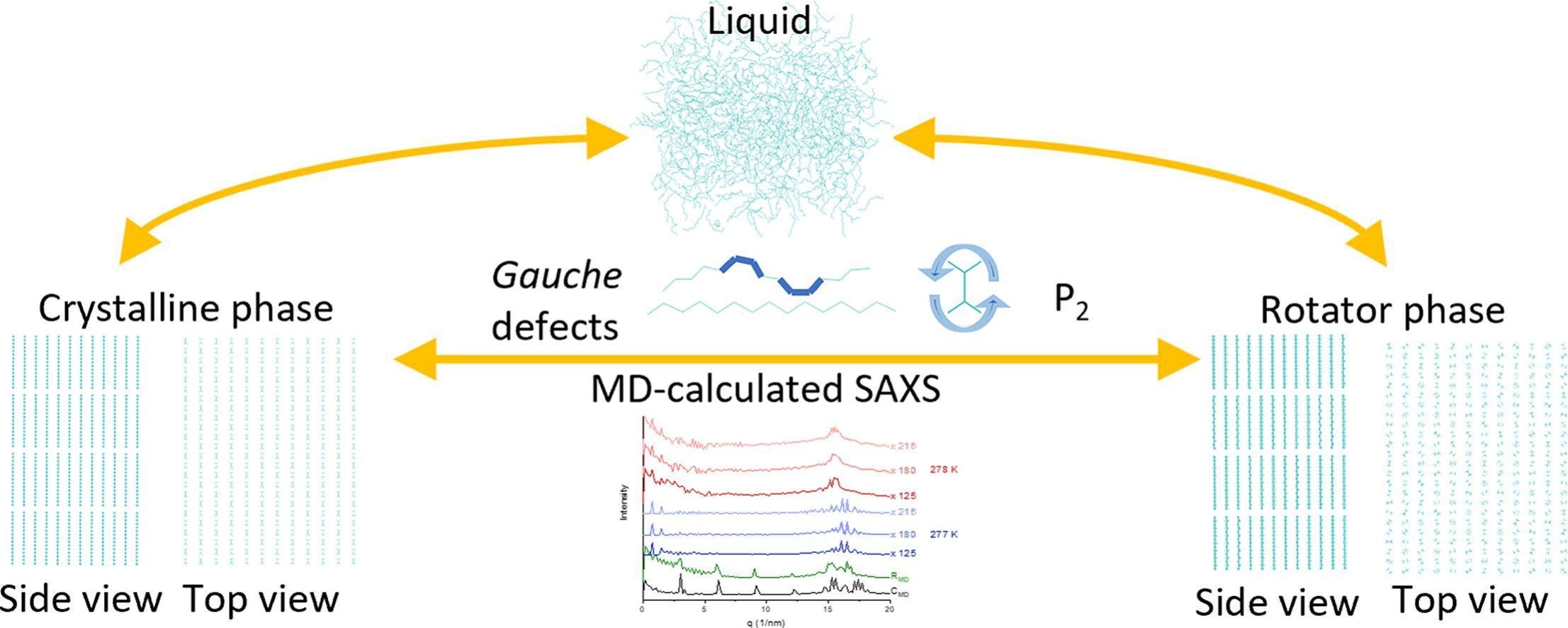
Computational freezing of pentadecane
Molecular dynamics simulations are employed to investigate the crystallization of pentadecane-containing systems. Reference crystalline and rotator phase constructed from crystallographic data benchmark the structures formed upon cooling. Phase identification is achieved through global and local structural descriptors, with the fraction of gauche conformations and angular P₂ profiles outlined as the most sensitive indicators.
Pentadecane exhibits a markedly more stable rotator phase than hexadecane. This is confirmed by simulations spanning more than 30 K for the reference rotator phase. In this temperature range, a model regular rotator phase of pentadecane remains stable without undergoing significant structural changes, showing high reproducibility across independent trajectories. This contrasts hexadecane, for which a rotator phase rapidly transforms toward a triclinic structure [Iliev et al. 2023]. The presence of a surfactant in the system promotes heterogeneous nucleation, shifts crystallization to higher temperatures, and stabilizes the rotator phase, in agreement with experiments.
A computationally efficient protocol for simulating SAXS spectra from MD trajectories is proposed. The simulated spectra align very well with experimental data for both crystalline and rotator phases. Crystallographic lattice parameters can be extracted from the most intense SAXS peaks, even for experimentally unknown structures, demonstrating the general applicability of the approach to solid-state phase analysis.
ISCOM-type matrix from beta-escin and glycyrrhizin saponins
Background and aims
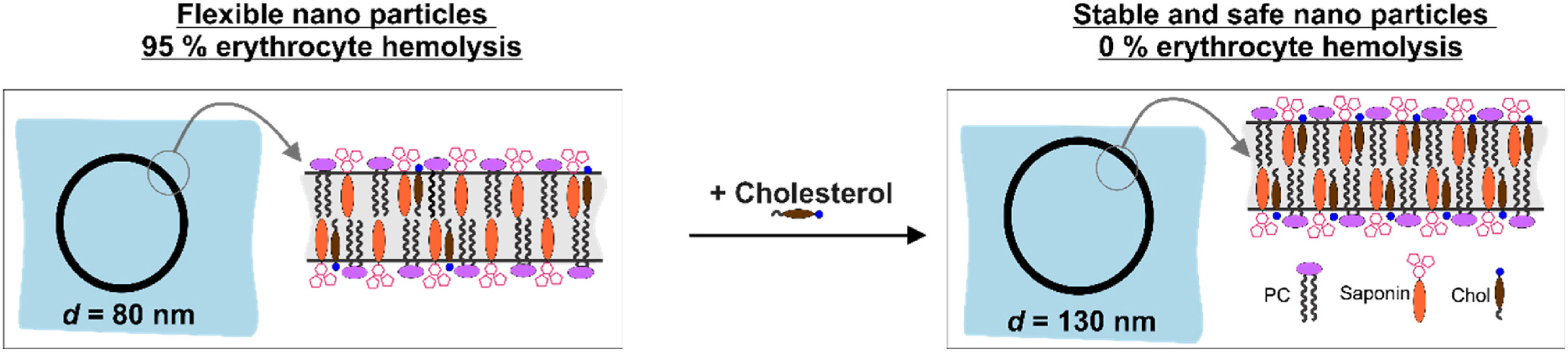
Nanotechnology provides the opportunity for construction of modern transport devices such as nanoparticles for a variety of applications in the field of medicine. A novel experimental protocol for the formation of saponin-cholesterol-phospholipid nanoparticles of vesicular structure has been developed and applied to prepare stable nanoparticles using escin or glycyrrhizin as saponins.
Methods
The methods for nanoparticle construction include a sonication at 90 °C of the initial mixture of components, followed by an additional sonication on the next day for incorporation of an additional amount of cholesterol, thus forming stable unilamellar vesicles. Tests and assays for cell viability, erythrocyte hemolysis, flow cytometry, and fluorescent microscopy analyses have been performed.
Results
By selecting appropriate component ratios, stable and safe particles were formulated with respect to the tested bio-cells. The prepared nanoparticles have mean diameter between 70 and 130 nm, depending on their composition. The versatility of these nanoparticles allows for the encapsulation of various molecules, either within the vesicle interior for water-soluble components or within the vesicle walls for hydrophobic components. The saponin particles formed after cholesterol post-addition (E3-M2) are stable and 100 % of the cells remain viable even after 10-times dilution of the initial particle suspension. These particles are successful included into isolated mouse macrophages.
Conclusions
Among the variety of generated nanoparticles, the E3-M2 particles demonstrated properties of safe and efficient devices for future vaccine design and antigen targeting to immune system.
Types of phases obtained by molecular dynamics simulations upon freezing of hexadecane-containing systems
Medium- to long-chain alkanes can form upon cooling intermediate phases between isotropic liquid and solid crystalline, called rotator phases, where relative freedom of the molecules to rotate about their long axis is combined with long range translational order. Rotator phases are well documented experimentally but the mechanism of their formation at the molecular level is still not fully explained. In a previous work, we have shown that molecular dynamics simulations can produce rotator phases upon cooling of hexadecane [S. Iliev et al., J. Col. Int. Sci., 2023, 638, 743]. The aim of the current work is to develop a procedure to identify the specific ordered phase obtained in the simulations. The influence of the cooling rate on the freezing process of hexadecane (bulk and surfactant-interfaced to water) is tested as well. Several parameters are combined to quantify the degree of ordering and the type of phase in the studied systems. These are the tilt angle of the molecules with respect to the crystallite plane, the radial distribution function of the centre of mass of the molecules in the crystallite, the percentage of the gauche torsion angles in the molecules, the angle of the second principal axis of each molecule with respect to the x axis of the coordinate system, and estimates from Voronoi analysis. The results show that the systems form a rotator phase, which transitions gradually towards the thermodynamically most stable triclinic crystal, and the transformation progresses to different extent depending on the system. The influence of the cooling rate is related only to the size of the largest crystallite formed, the other parameters of the freezing process remain unaffected. The work also presents a robust procedure for obtaining and identifying different types of ordered phases in alkane-containing systems with thoroughly tested computational protocol and a comprehensive set of structural analyses. Several key characteristics are advanced, compared to previous research [Ryckaert et al., Mol. Phys., 1989, 67, 957; Wentzel et al., J. Chem. Phys. 2011, 134, 224504], namely, a new methodology is proposed to compute the unit cell deformation parameter and azimuthal angle from MD simulation trajectories of the freezing process in alkane-containing systems. The suggested structural analysis, which is independent of the coordinate system, is applicable to any linear-chain system with polycrystalline structure.

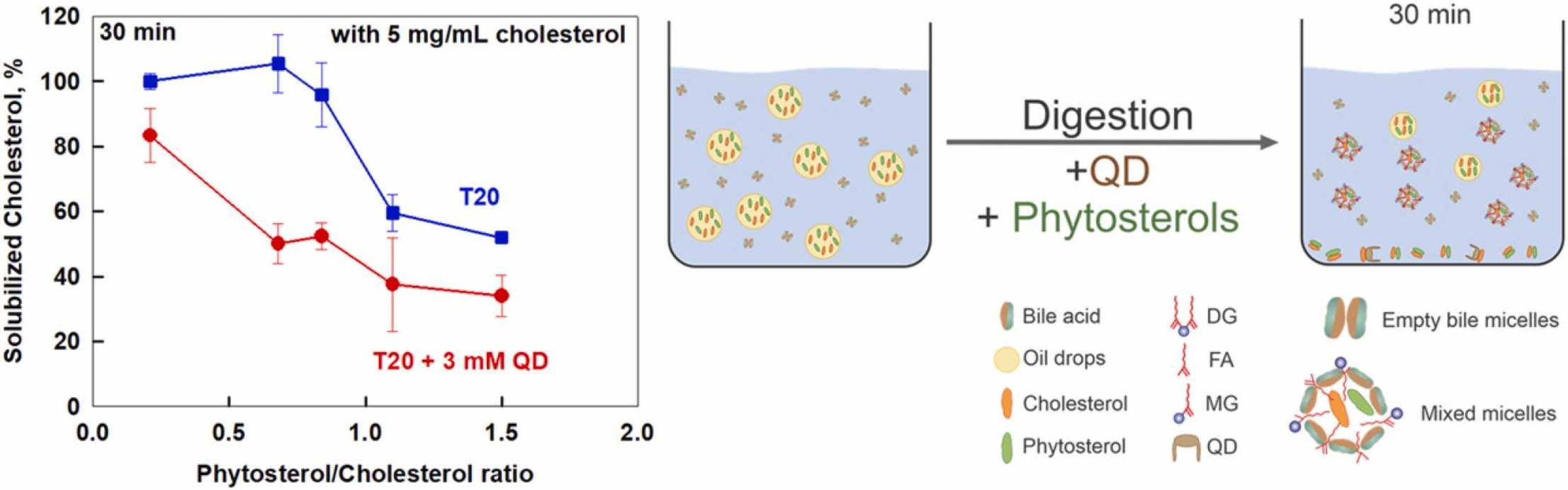
Cholesterol solubilization: Interplay between phytosterols, saponins and lipid digestion products
High plasma concentrations of cholesterol are associated with cardio-vascular disease complications and high risk of myocardial infarction. The absorption of dietary cholesterol depends on its bioaccessibility, which is in turn influenced by phytosterols, saponins and lipid digestion products. Therefore, we explored the interplay between phytosterols, Quillaja Dry saponin extract (QD), their combinations and lipid digestion on cholesterol bioaccessibility via an in vitro.
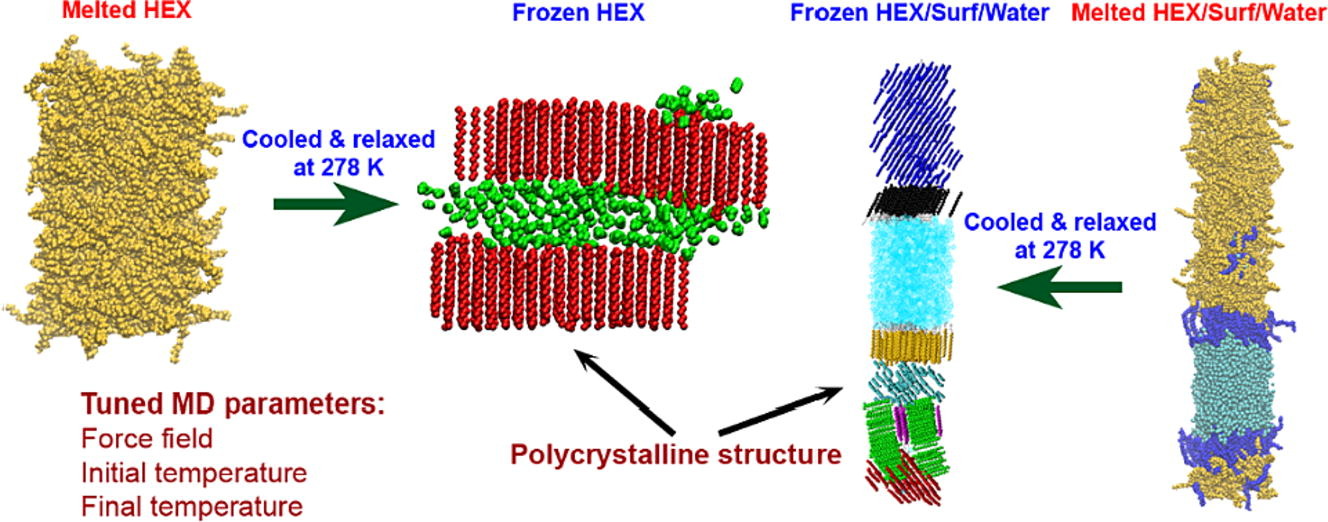
Computational assessment of hexadecane freezing by equilibrium atomistic molecular dynamics simulations
Hypothesis: Upon cooling, alkanes can form intermediate phases between liquid and crystal. They are called “rotator” or “plastic” phases and have long-range positional order with rotational freedom around the long molecular axis which gives them non-trivial and useful visco-plastic properties. We expect that the formation and structure of rotator phases formed in freezing alkanes can be understood much deeper by tracking the process at molecular level with atomistic molecular dynamics. Simulations: We defined an appropriate CHARMM36-based computational protocol for simulating the freezing of hexadecane, which contained a sufficiently long (500 ns) equilibrium sampling of the frozen states. We employed it to simulate successfully the freezing of bulk and interface-contacting hexadecane and to provide a pioneering clarification of the effect of surfactant on the crystallization mechanism and on the type of intermolecular ordering in the crystallites. Findings: The devised computational protocol was able to reproduce the experimentally observed polycrystalline structure formed upon cooling. However, different crystallization mechanisms were established for the two types of models. Crystallites nucleate at random locations in the bulk and start growing rapidly within tens of nanoseconds. In contrast, the surfactants freeze first during the fast cooling (<1 ns), followed by rapid hexadecane freezing, with nucleation starting along the entire surfactant adsorption layer. Thereby, the hexadecane molecules form rotator phases which transition into a more stable ordered phase. This collective transition is first-time visualized directly. The developed robust computational protocol creates a foundation for future in-depth modelling and analysis of solid-state alkane-containing, incl. lipid, structures.