Prof. Slavka S. Tcholakova, Ph.D.
Head of the Laboratory for Active Formulations and Materials
Interests
- Formation, stability and rheology of foams and emulsions
- Mechanism of action of antifoams
- Surfactant aggregates and mesophases in bulk: structure, rheology, stability
- Biophysics & colloid science in fat digestion and drug delivery systems
- Defoamers and antifoams
- Physicochemical theoretical models for foamability, emulsification; foam rheology
- Natural based surfactants
Bio
Prof. Slavka Tcholakova received M.Sc. in Chemistry (1996), Ph.D. in Physical Chemistry (2004), Assistant Professor (2006), Associate Professor (2009), and became a Full Professor in 2013 in the Faculty of Chemistry and Pharmacy, Sofia University, Bulgaria. She was a head of the Department of Chemical and Pharmaceutical Engineering (DCPE) in Sofia University from 2015 to 2024. Currently, she is a head of the Laboratory “Active formulations and materials” at DCPE. She has been a visiting researcher in the Research Center Paul Pascal, CNRS, Bordeaux, France (1997).
Her research interests include formation and stability of emulsions; rheology of foams and emulsions; protein adsorption in relation to emulsion stability; foam stability in the presence of antifoams; in-vitro models for digestion and bioavailability of hydrophobic molecules and the respective experimental methods. So far, she has published 150 research and review papers, cited over 6300 times (h-index = 44). She has been leading over 70 projects and participating in more than 30 projects with international companies (BASF, Unilever, Saint Gobain, Wacker, Lubrizol, PepsiCo, Altana, BYK Chemie, Productolysa, etc.).
She has been a supervisor and co-supervisor of 16 completed PhD Theses. She was the recipient of the “Best Young Scientist” award for 2006 of the Sofia University Foundation “St. Kliment Ohridski”. In 2018, Prof. Tcholakova received the National Bulgarian award “Pythagoras” for high scientific achievements in category Natural Sciences.
Based on her publications and citations in 2023 and 2024, Prof. Tcholakova achieved high rankings in the Chemical Physics category of the Stanford/Elsevier’s Top 2% Scientists Ranking: In 2024, she was ranked 1712 out of 106,831 scientists; In 2025, she was ranked 1764 out of 115,551 scientists.
Publications
Most recent publications
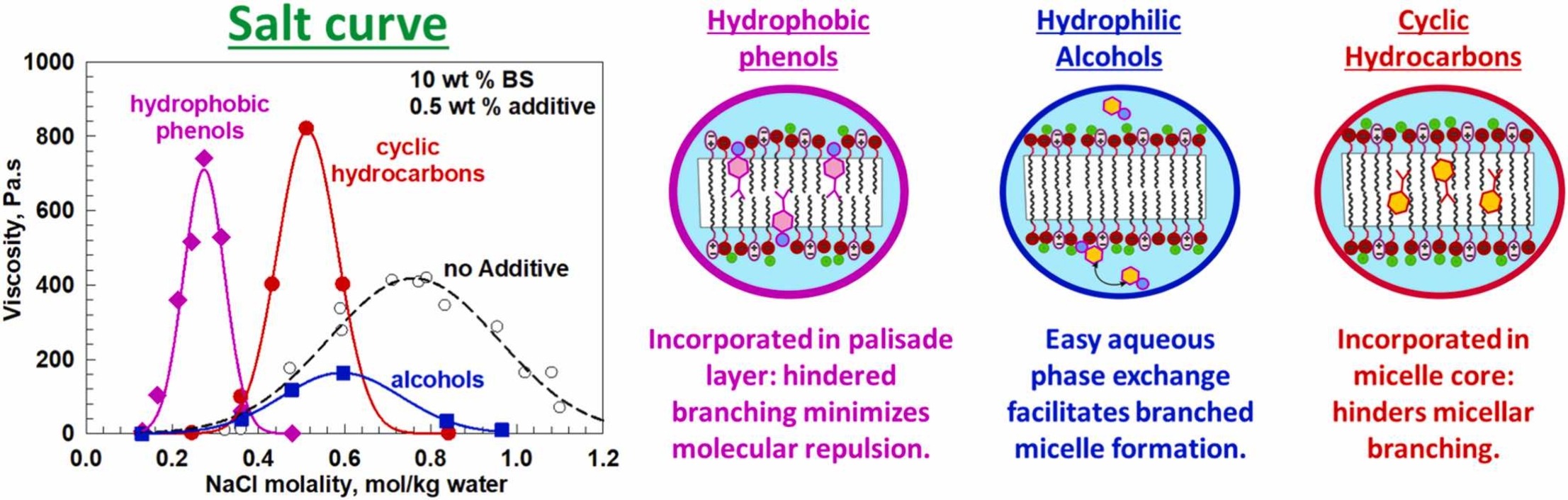
Effect of cyclic molecules on salt curves of mixed anionic-zwitterionic surfactant solutions
The effect of twelve cyclic molecules on the rheological response of a 10 wt% mixture of sodium lauryl ether sulfate and cocoamidopropyl betaine (BS) was evaluated. The results demonstrate that all studied additives effectively reduced the salt concentration required to reach peak viscosity and narrowed the width of the salt curve. The specific effect on the magnitude of the peak viscosity depends on the molecular structure of the additive. Hydrocarbons, along with hydrophobic phenols, increase the peak viscosity, while alcohols and simple phenol, decrease it. SAXS and NMR measurements revealed that hydrocarbons are primarily incorporated into the micellar core, and the observed viscosity enhancement is attributed to the suppression of micellar branching. In contrast, molecules possessing an OH-group intercalate between the surfactant headgroups or reside at the micellar surface. For hydrophobic phenols, the ability to form stable hydrogen bonds results in a significant increase in the maximum viscosity. This interaction substantially reduces the salt concentration required to reach peak viscosity; in this case, the micellar charge density remains relatively high, and electrostatic repulsion hinders micellar branching. On the other hand, simple phenols and alcohols decrease viscosity because their higher water solubility allows them to easily redistribute across different regions of the micelle surface, thereby facilitating micellar branching. The dimensionless parameters accounting for the effect of the studied additives on the salt curve characteristics were determined. These parameters were shown to depend on a composite molecular parameter, defined as the ratio of the surfactant volume to the additive volume multiplied by the octanol–water partition coefficient, vBS/vALogP.
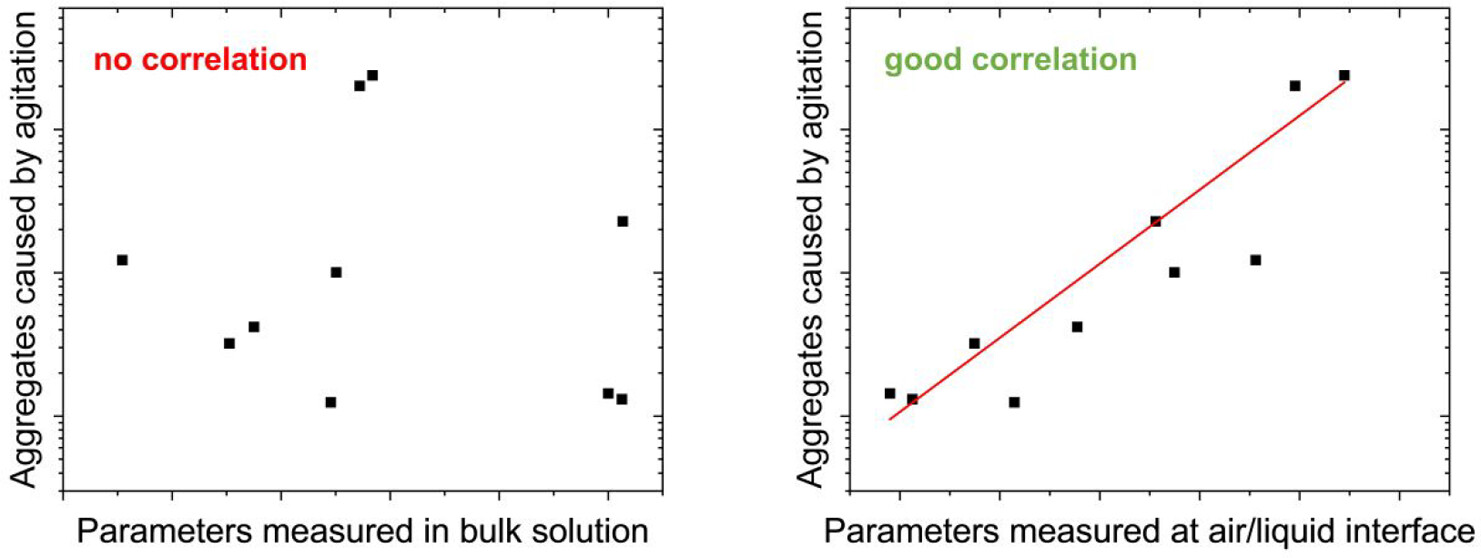
Predicting agitation stability of monoclonal antibodies during developability assessment
Developability assessment facilitates the selection of antibody drug candidates with desirable pharmaceutical properties. However, it remains uncertain whether agitation-induced aggregation can be predicted from standard developability parameters. Here, we investigated whether key biophysical parameters predict agitation-induced aggregation of monoclonal antibodies (mAbs). To this end, we generated a benchmark data set by characterizing the aggregation upon agitation in the presence of an air–liquid interface of ten approved mAbs reformulated in a common surfactant-free buffer. The extent of aggregation varied substantially among mAbs and was primarily dependent on antibody identity. Flow imaging microscopy combined with machine learning revealed micrometre-sized aggregates with distinct morphologies, consistent with aggregation at air–liquid interfaces. Examination of thin liquid films and foams confirmed the presence of aggregates directly at the air–liquid interface and, therefore, the critical role of this interface for antibody aggregation during agitation. We then applied fluorescence-based, light scattering, and chromatographic techniques to determine standard developability parameters for each mAb, including apparent melting temperature (Tm), nonreversibility onset temperature (Tnr), aggregation onset temperature (Tagg), diffusion self-interaction parameter (kD), hydrophobic interaction chromatography retention time, and relative monomer yield after isothermal refolding from chemical denaturants. Notably, none of these parameters correlated with agitation-induced aggregation. Finally, we assessed the surface properties of the mAbs via drop shape analysis and found that the combination of surface pressure and elastic modulus yields a good correlation with the concentration of micrometre-sized aggregates formed due to agitation. Overall, these findings highlight limitations in predicting mAb interfacial stability using standard developability assays and underscore the importance of studying antibody behavior at interfaces.
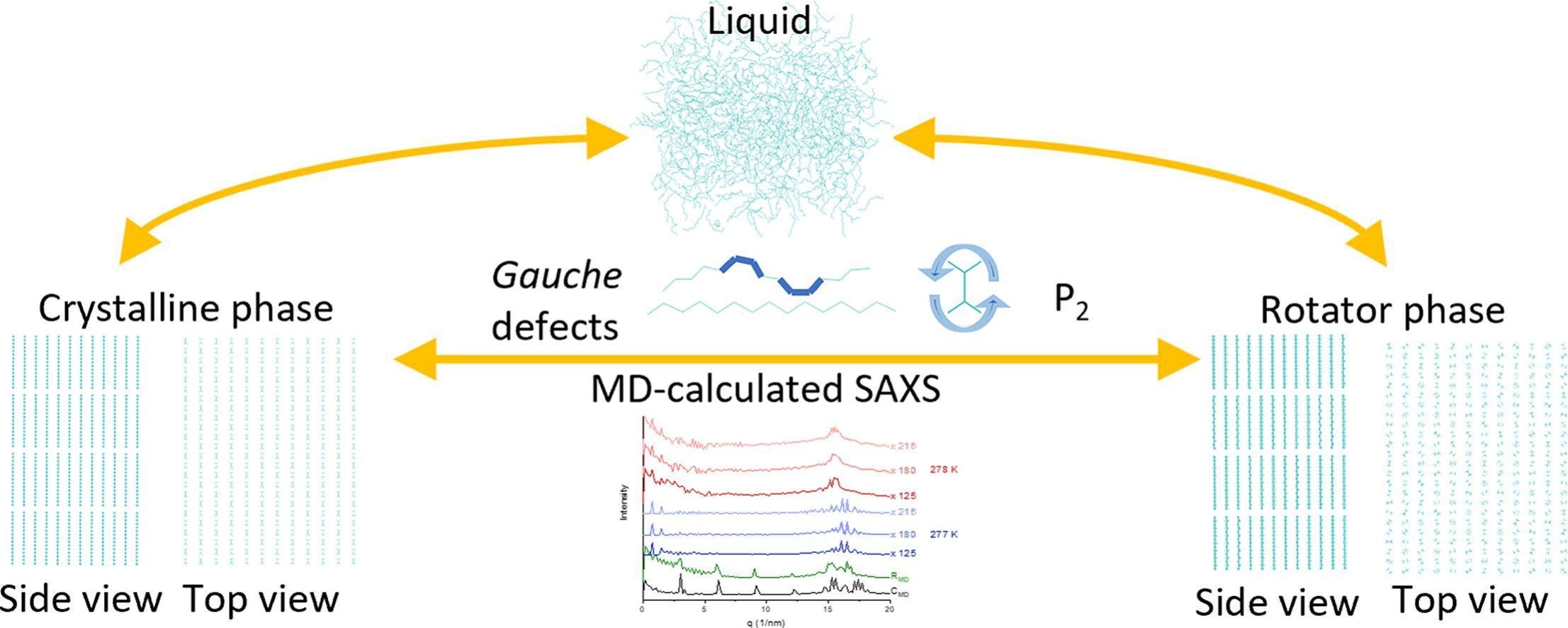
Computational freezing of pentadecane
Molecular dynamics simulations are employed to investigate the crystallization of pentadecane-containing systems. Reference crystalline and rotator phase constructed from crystallographic data benchmark the structures formed upon cooling. Phase identification is achieved through global and local structural descriptors, with the fraction of gauche conformations and angular P₂ profiles outlined as the most sensitive indicators.
Pentadecane exhibits a markedly more stable rotator phase than hexadecane. This is confirmed by simulations spanning more than 30 K for the reference rotator phase. In this temperature range, a model regular rotator phase of pentadecane remains stable without undergoing significant structural changes, showing high reproducibility across independent trajectories. This contrasts hexadecane, for which a rotator phase rapidly transforms toward a triclinic structure [Iliev et al. 2023]. The presence of a surfactant in the system promotes heterogeneous nucleation, shifts crystallization to higher temperatures, and stabilizes the rotator phase, in agreement with experiments.
A computationally efficient protocol for simulating SAXS spectra from MD trajectories is proposed. The simulated spectra align very well with experimental data for both crystalline and rotator phases. Crystallographic lattice parameters can be extracted from the most intense SAXS peaks, even for experimentally unknown structures, demonstrating the general applicability of the approach to solid-state phase analysis.
Drug solubilization in simulated intestinal fluids vs lipophilicity: Does charge matter?
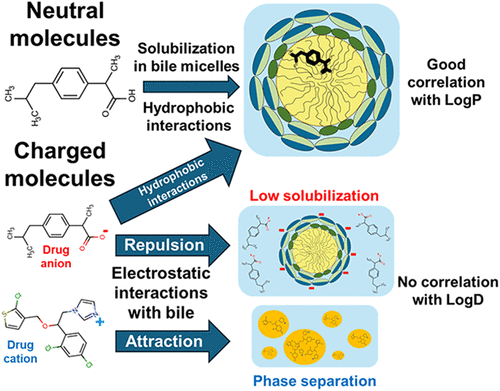
Solubilization of poorly water-soluble drugs in human intestinal fluids influences oral absorption and is linked to food effects. Current empirical equations for calculating intestinal solubilization via lipophilicity are built on limited data and do not adequately account for drug ionization. We aim to expand the data set and build a model to clarify the link between lipophilicity and solubilization for charged compounds. We determined the aqueous solubility, octanol–water partition coefficient, and solubilization in fed-state simulated intestinal fluids (FeSSIF) of 26 hydrophobic drugs. Combined with literature data, a good correlation (R2 = 0.74, n = 198) between intestinal solubilization and LogP/D was observed. However, data segregation showed that the solubilization of neutral compounds correlated very well with LogP (R2 = 0.89, n = 114), whereas the correlation with LogD was lost for the charged compounds (R2 = 0.40, n = 84). To better understand this behavior, the pH of FeSSIF was varied to study the solubilization of the same compounds in the neutral and charged states. While a very good correlation between solubilization and LogD was observed in the neutral state of the compounds (R2 = 0.92, n = 8), the correlation was again lost (R2 = 0.02, n = 4) in their charged state. Electrostatic interactions were suggested to play a key role in the unexpectedly low solubilization of anionic drugs and in the phase separation observed for cationic drugs. The presented insights further advance the understanding of the solubilization of hydrophobic drugs in biorelevant media and provide a foundation for broader and improved modeling approaches.
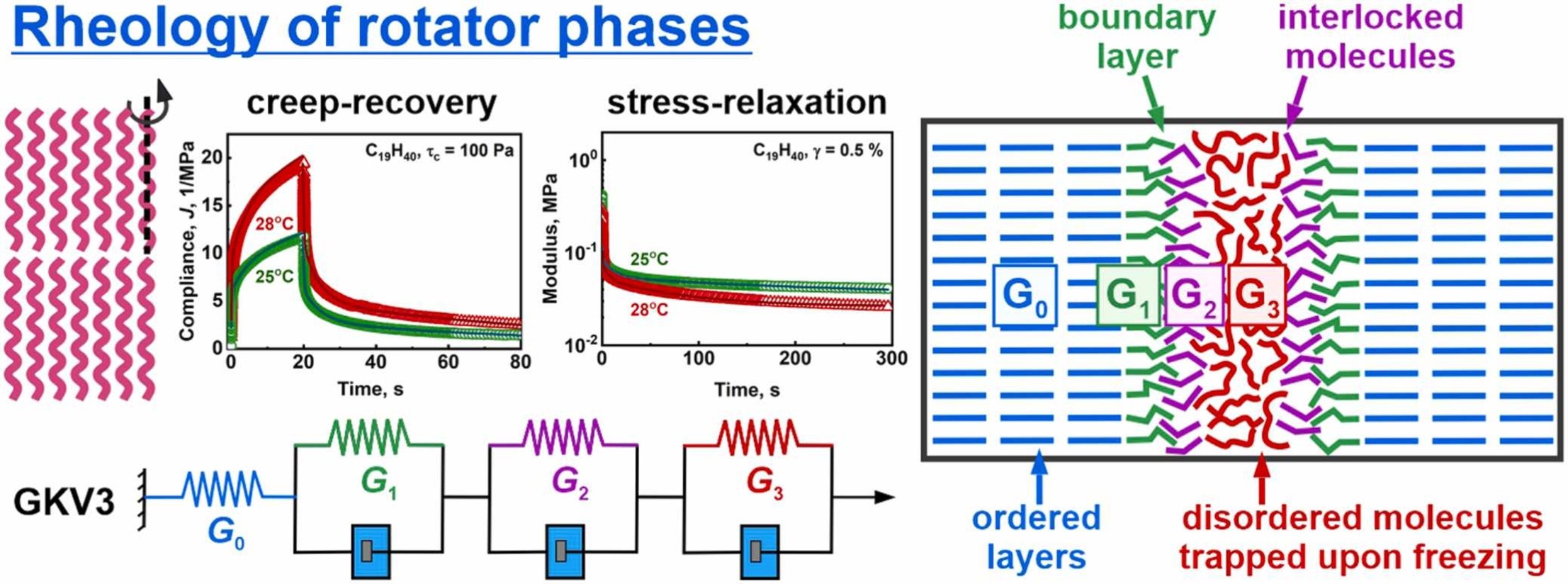
Rheological characterization of solid lipids with domain structure
Phase behavior of lipids is of primary importance for the manufacturing and applications of foods, cosmetics and pharmaceuticals, as well as for the functions of biological membranes. Upon cooling, the molten bulk lipids crystallize into ordered domains which determine their rheological properties. While storage and loss moduli are typically used to describe these properties, their direct connection to the underlying molecular rearrangement remains poorly understood. In the current study, we performed a detailed rheological characterization of the rotator phases (intermediate phases between fully ordered crystalline and completely disordered liquid phases) formed in bulk linear alkanes. Large series of stress-relaxation and creep-recovery experiments were performed and interpreted, using generalized Kelvin-Voigt model with one spring, connected in series with three combined elements of a spring and a dashpot. We determined the elasticities and viscosities of all these rheological elements, along with the respective three relaxation times of the combined elements. These relaxation times are governed by different molecular processes in the sheared samples and differ by three orders of magnitude: t1 ≈ 0.45 s and t2 ≈ 8–9 s are related to local molecular rearrangements at the domain boundaries, while t3 ≈ 140–200 s most probably describes the rearrangement of disordered lipid molecules entrapped between the ordered domains. The storage and loss moduli, calculated from the constants of the generalized Kelvin-Voigt model, were in a very good agreement with those measured directly in amplitude sweep and temperature ramps oscillatory tests, thus supporting the self-consistency of data interpretation. The methodology presented here is applicable to other polycrystalline lipid materials with 2D or 3D domain structures, providing a valuable framework for interpreting their rheological behavior.