Computational freezing of pentadecane
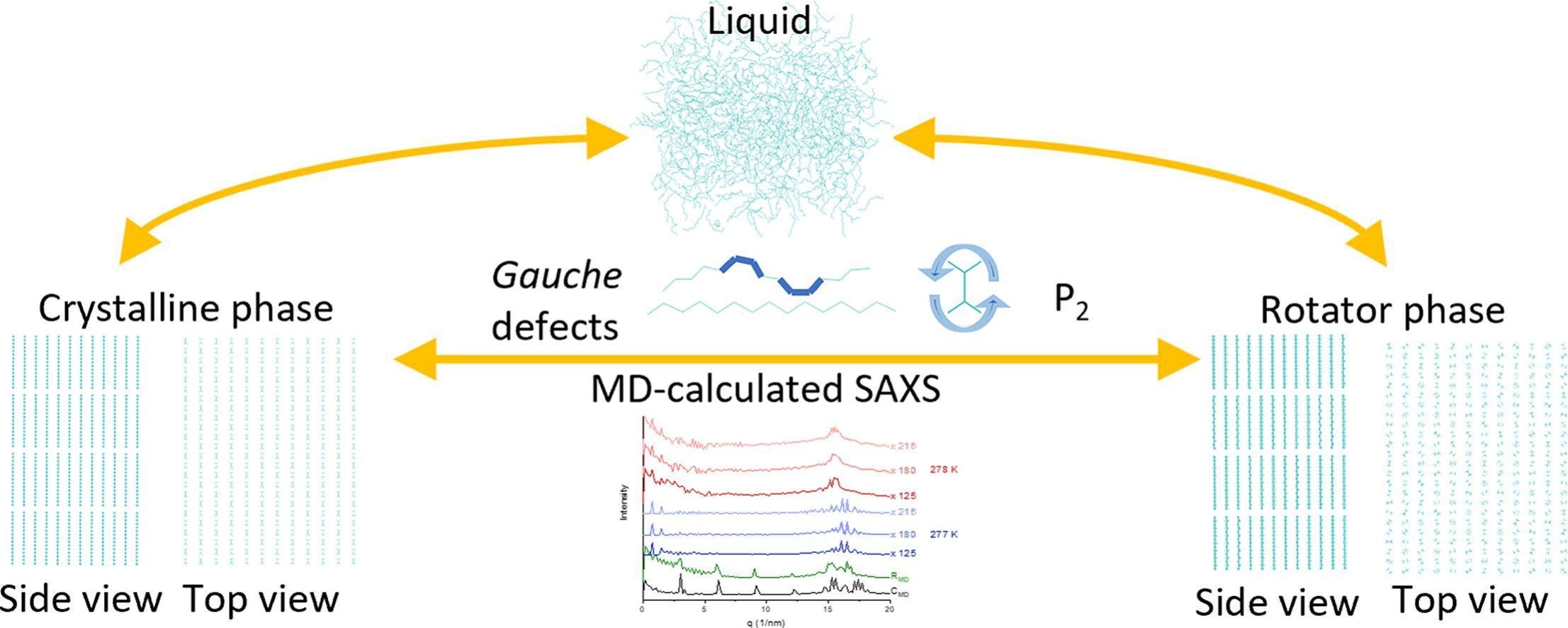
Molecular dynamics simulations are employed to investigate the crystallization of pentadecane-containing systems. Reference crystalline and rotator phase constructed from crystallographic data benchmark the structures formed upon cooling. Phase identification is achieved through global and local structural descriptors, with the fraction of gauche conformations and angular P₂ profiles outlined as the most sensitive indicators.
Pentadecane exhibits a markedly more stable rotator phase than hexadecane. This is confirmed by simulations spanning more than 30 K for the reference rotator phase. In this temperature range, a model regular rotator phase of pentadecane remains stable without undergoing significant structural changes, showing high reproducibility across independent trajectories. This contrasts hexadecane, for which a rotator phase rapidly transforms toward a triclinic structure [Iliev et al. 2023]. The presence of a surfactant in the system promotes heterogeneous nucleation, shifts crystallization to higher temperatures, and stabilizes the rotator phase, in agreement with experiments.
A computationally efficient protocol for simulating SAXS spectra from MD trajectories is proposed. The simulated spectra align very well with experimental data for both crystalline and rotator phases. Crystallographic lattice parameters can be extracted from the most intense SAXS peaks, even for experimentally unknown structures, demonstrating the general applicability of the approach to solid-state phase analysis.