Computational assessment of hexadecane freezing by equilibrium atomistic molecular dynamics simulations
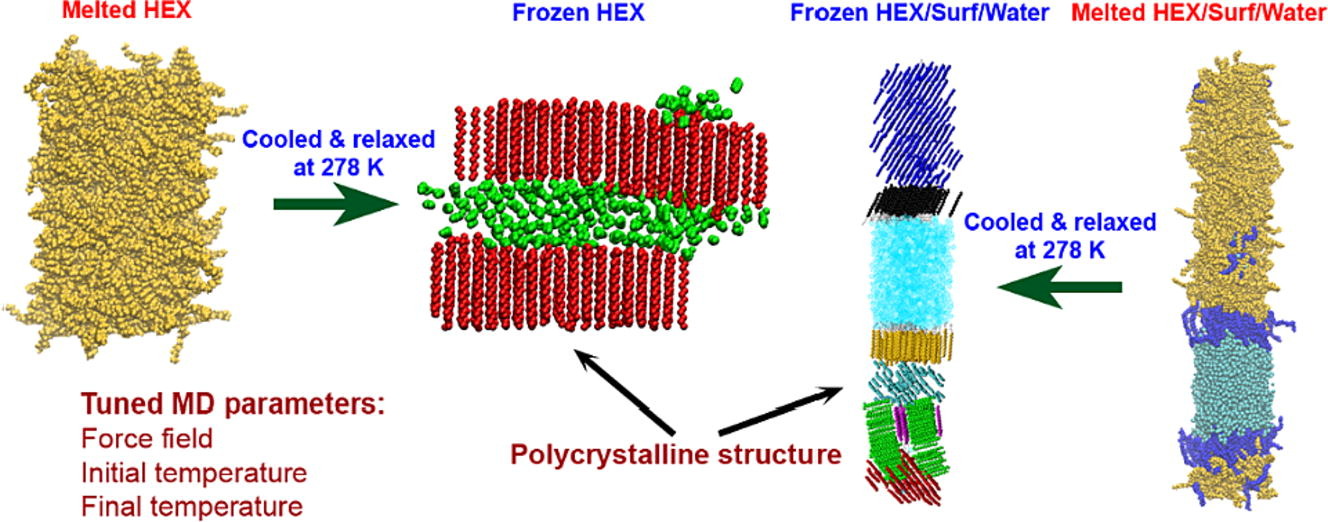
Hypothesis: Upon cooling, alkanes can form intermediate phases between liquid and crystal. They are called “rotator” or “plastic” phases and have long-range positional order with rotational freedom around the long molecular axis which gives them non-trivial and useful visco-plastic properties. We expect that the formation and structure of rotator phases formed in freezing alkanes can be understood much deeper by tracking the process at molecular level with atomistic molecular dynamics. Simulations: We defined an appropriate CHARMM36-based computational protocol for simulating the freezing of hexadecane, which contained a sufficiently long (500 ns) equilibrium sampling of the frozen states. We employed it to simulate successfully the freezing of bulk and interface-contacting hexadecane and to provide a pioneering clarification of the effect of surfactant on the crystallization mechanism and on the type of intermolecular ordering in the crystallites. Findings: The devised computational protocol was able to reproduce the experimentally observed polycrystalline structure formed upon cooling. However, different crystallization mechanisms were established for the two types of models. Crystallites nucleate at random locations in the bulk and start growing rapidly within tens of nanoseconds. In contrast, the surfactants freeze first during the fast cooling (<1 ns), followed by rapid hexadecane freezing, with nucleation starting along the entire surfactant adsorption layer. Thereby, the hexadecane molecules form rotator phases which transition into a more stable ordered phase. This collective transition is first-time visualized directly. The developed robust computational protocol creates a foundation for future in-depth modelling and analysis of solid-state alkane-containing, incl. lipid, structures.